Article Text

Statistics from Altmetric.com
According to the most recent definition, bronchial asthma is a chronic inflammatory disorder of the airways associated with reversible airway obstruction and increased airway responsiveness to a variety of stimuli.1 An intuitive inference from this definition is that a causal relationship may exist between airway inflammation and airway hyperresponsiveness. Along this line of reasoning, most of the research in the last two decades in this field was aimed at identifying inflammatory cell products possibly responsible for the pathogenesis of bronchial asthma and airway hyperresponsiveness.2 However, the common observation that the asthmatic airways are equally hyperresponsive to a variety of different stimuli does challenge the idea that a single inflammatory cell or mediator may be central to the pathogenesis of asthma and airway hyperresponsiveness and focuses on the importance of an altered mechanical response of the target organ. This view has been recently corroborated by the finding that airway responsiveness of normal individuals may become similar to that of asthmatics by simply changing the pattern of breathing during the bronchial challenge.3It is therefore legitimate to wonder how much of airway hyperresponsiveness is due to inflammation or to inherent predisposing factors.
The aims of the present review are to show that airway narrowing in asthma is the ultimate result of an interaction between complex and multiple mechanisms not necessarily and uniquely related to airway inflammation, and to revisit the evidence on which the theorem “airway inflammation equal to airway hyperresponsiveness” has been constructed.
Mechanisms of airway narrowing in asthma
Airway calibre in vivo is the result of a delicate balance between the force generated by the airway smooth muscle (ASM) and a number of opposing factors.4 The latter are mainly represented by autonomic mechanisms that tend to limit the ASM tone and by mechanical forces that oppose ASM shortening.
AIRWAY SMOOTH MUSCLE (ASM)
Although it is widely accepted that ASM contraction is a major cause for airway obstruction in asthma, there is uncertainty as to whether abnormalities of ASM play any part in the pathogenesis of airway hyperresponsiveness. In terms of force-length characteristics, the asthmatic ASM does not behave differently from the normal ASM.5 ,6 These data lend support to a purely immunological view of bronchial asthma in which the ASM cell is regarded as a normal effector of response to abnormal inflammatory stimuli. Nevertheless, the smooth muscle of asthmatic airways may be able to generate greater force than normal because of hypertrophy and hyperplasia,7thus causing excessive airway narrowing.8 ,9 Although the thickness of the ASM in the airway wall of asthmatic patients might have been overestimated10 and there is no proof that an increased mass of smooth muscle is associated with an increased force generation capacity,11 the possibility that an increased contractile force developed by ASM might contribute to airway hyperresponsiveness in asthma cannot be ruled out.
Recent work has suggested that normal and asthmatic ASM may differ in terms of length shortening characteristics. The capacity and the velocity of shortening are increased after passive sensitisation of ASM.12 The increase of shortening velocity is due to an increase in the amount and the activity of the myosin light chain kinase13 and increased phosphorylation of the 20 kD regulatory unit with increased cross-bridge cycling rate.14-17 Albeit small, the difference in shortening velocity between asthmatic and normal ASM may be relevant to the generation and maintenance of force. Under normal conditions stretching causes a transient reduction in ASM tone.18 ,19 This is considered an important mechanism for preventing excessive airway narrowing (see below) but it may be less efficient if the velocity of shortening is increased.20
Moreover, sensitised ASM may develop a myogenic contractile response to stretching.21 This response, which is believed to be a consequence of conversion from multi-unit to single-unit ASM,22 has been invoked to explain the sustained bronchoconstriction that occurs in some asthmatics after taking one or more deep breaths.23 ,24
MECHANICAL AND GEOMETRIC FACTORS
The mechanical properties of ASM are such that, if unimpeded, it may shorten to about 20–30% of its initial length when an appropriate stimulus is applied.25 In vivo, such a shortening would result in complete airway closure.8 In normal humans the maximal response to bronchoconstrictor stimuli is limited,26 which suggests that some mechanisms opposing ASM shortening are operative in vivo.
In vitro the shortening of ASM is considerably less when elastic loads are applied.27 Furthermore, human ASM shortens less than ASM from other species and this difference seems to be related to a greater amount of connective tissue present in human bronchi, which represents a parallel elastic load.28 It has been suggested that a decrease in the elastic load internal to ASM may contribute to greater ASM shortening in asthma.29 The cartilaginous rings and plates may also limit ASM shortening, thus preventing excessive airway narrowing8 ,28; in rabbits, softening the tracheal cartilage by papain consistently increases airway resistance.30
The most efficient force opposing ASM shortening in vivo is provided by the elastic recoil of the lung.8 ,28 When this is reduced, as in emphysema, the airways can become more narrow more easily.31 Although there is no consistent proof that lung elastic recoil is reduced in asthma,32-35 even small decrements of transpulmonary pressure near functional residual capacity may have a deleterious effect when the ASM contracts. In normal individuals the response to methacholine is greatly enhanced by breathing just 500 ml below FRC—that is, at a lung volume at which transpulmonary pressure is only a few cm H2O less.36
The load imposed on the airways by the surrounding lung parenchyma changes during breathing. Due to their visco-plasto-elastic properties, both lung parenchyma and airways dissipate energy during breathing.18 ,19 ,37 ,38 For example, the force necessary to elongate the ASM in vitro is slightly but consistently greater than that necessary to bring it back to its original length.18 ,19 ,38 Such an energy dissipation becomes as much as 15 times greater when the ASM is contracted, depending on the frequency and amplitude of the length oscillations.38Basically, energy dissipation implies less ASM force and, by inference, less bronchoconstriction. During inspiration the airways are stretched by the force of interdependence through which they are coupled with lung parenchyma. If this force is effectively applied to the external airway wall, then ASM shortening may be opposed. Increasing tidal volume therefore reduces the response to bronchoconstrictor stimuli.39 ,40 When a stretch of similar amplitude of a deep inspiration to total lung capacity is applied, ASM is fully elongated and most of the energy generated during active shortening is dissipated.38 ,39 In normal individuals a deep inspiration is able to reverse a bronchoconstriction fully, enough to cause as much as a 50% decrease in forced expiratory flow.32 ,33 ,41-44 In asthmatic subjects deep inspiration has less bronchodilator effect on constricted airways32 ,33 ,42 ,44-46 or may even cause bronchoconstriction.46 ,47 The importance of the ability of deep inspiration to dilate the airways has recently been explored by Skloot et al 3 and by Pellegrino et al.32 In normal individuals prevention of deep inspirations during a bronchial challenge causes dyspnoea and the airway responsiveness becomes similar to that of asthmatics.8 ,32 An inference from these data may be that airway hyperresponsiveness is more a problem of inability to dilate constricted airways rather than of increased constrictor stimuli to ASM.
In a simple model where airway narrowing is uniquely due to ASM shortening, the contractile elements may return to their relaxed status after being stretched because the actin-myosin cross bridges are detached. Then, the airways remain dilated after a deep inspiration until cross bridges reform and the tone prior to the deep inspiration is re-established.39 This is what seems to occur in normal individuals during induced bronchoconstriction.41-44 To understand why the bronchodilator response to deep inspiration in asthmatic subjects is blunted or even reversed, a more complex model must be invoked where ASM does not dissipate energy during cycling and/or it is prevented from doing so by external factors. For the first condition to be true, primary defects of the ASM that prevent detachment of cross bridges must be present. To date there are no data to support such a mechanism, which suggests that the behaviour of the ASM is more likely to be regulated by external forces. In asthma the airways may be less sensitive to the action of external forces because the force of interdependence is reduced3 ,4 ,33 ,36 ,41 ,46 ,47 or the non-contractile elements in the airway wall are more stiff.17 According to Fredberg et al 17 the cross bridge cycling rate decreases if the ASM stretching is impeded, and the conversion to slow cycling latch bridges occurs which promotes maintenance of steady state tone and increased stiffness of the ASM. Thus, even a minimal transient unloading of the ASM due to the effects of deep inspiration on the non-contractile tissues external to the ASM—for example, the lung or parenchyma—could result in passive shortening of the ASM. This could explain, at least in part, the blunted bronchodilator effect of deep inspirations associated with the increase in parenchymal hysteresis in asthma.46 Furthermore, if the velocity of shortening of the ASM in asthma is increased,12-17 the active force may be quickly re-established after inspiration so that the ASM is rapidly unloaded upon expiration.20 Recently, Gunst et al 48 advanced the hypothesis that the force developed by the ASM in response to constrictor stimuli depends on the length at which the stimulus is applied. If the ASM contracts at longer length and then is shortened, the tensile force is less than if the ASM is contracted at shorter length. It has been suggested that this behaviour reflects a different arrangement of the contractile elements inside the ASM cell.40
The lack of protection from excessive airway narrowing observed in asthma can be, at least in part, reasonably attributed to airway inflammation or inflammatory remodelling of the airway walls.2 ,49 ,50 It has been suggested that peribronchial oedema may attenuate the pulling effect of parenchymal attachments on airway walls, thus unloading the ASM and allowing more shortening for the same intensity of stimulus.4 The airway wall thickening that occurs in bronchial asthma49 ,51 due to oedema, cellular infiltration, and vascular engorgement may amplify the bronchoconstrictor response as it causes airway calibre to decrease more for a given ASM shortening.52 However, the effect of mucosal thickening on airway narrowing is complex. Airway mucosa folds when ASM shortens,9 ,53 a process that requires energy dissipation and hence may represent a load for the ASM. In addition, the subepithelial fibrosis characteristic of asthma makes the airway stiff, thus opposing ASM shortening.54 On the other hand, the degree of airway narrowing is inversely proportional to the number of mucosal folds,53 which decreases with mucosal and basement membrane thickening.55 Paradoxically, the same inflammatory abnormalities could either favour or limit airway narrowing, depending on interactions with other concomitant changes.
In conclusion, owing to the complexity and multiple interactions of the mechanism regulating airway narrowing, it would be simplistic to regard airway inflammation as the only cause of airway hyperresponsiveness.
Airway inflammation and airway hyperresponsiveness
The concept of asthma as an inflammatory disorder derives from necroscopic studies on asthmatic subjects who died because of an asthma attack or other causes51 ,56 ,57 and in vivo studies using bronchoalveolar lavage, induced sputum, and bronchial biopsy specimens in subjects with asthma of different severity, either at baseline58-60 or after exposure to allergens or occupational sensitisers.61-63
The most prominent feature in necropsies of patients with fatal asthma is a marked thickening of airway walls. This is commonly associated with epithelial damage, thickening of basement membrane, marked increase of bronchial capillary bed, fluid exudation with oedema, goblet cell hyperplasia, ASM hyperplasia and hypertrophy, and intraluminal mucus and cellular debris causing complete or partial airway occlusion. The eosinophil is generally the dominating inflammatory cell in both fatal and non-fatal asthma64 ,65but neutrophils may prevail in cases of sudden onset fatal asthma.64 The airway walls of subjects with non-fatal asthma are also thicker than normal but to a lesser extent than those of subjects with fatal asthma.66 ,67
Common findings in the biopsy specimens of asthmatic airways are epithelial shedding, collagen deposition below the basement membrane,68 ,69 increased numbers of eosinophils and mast cells in the mucosa. Eosinophils, mast cells and their products are also increased in the bronchoalveolar lavage fluid.70 In this scenario the recruitment and activation of Th2 cells seem to play a central role.71
AIRWAY INFLAMMATION AS A CAUSE OF AIRWAY HYPERRESPONSIVENESS: PROS
In asthmatic subjects inhalation of allergen or occupational sensitisers causes an inflammatory (mainly eosinophilic) response in the airways which is associated with an increase in airway hyperresponsiveness.61 ,72 In both healthy humans and animals exposure to ozone causes an inflammatory (mainly neutrophilic) response and airway hyperresponsiveness.73 ,74 A temporal relationship exists between the inflammatory events occurring in the airways and the increase in airway responsiveness after exposure to allergens. Both the influx of eosinophils and the increase in airway responsiveness precede the development of the late phase response.75 ,76
Airways hyperresponsiveness often first appears or worsens following a viral infection of the upper airways or vaccination with live influenza virus. The increase in airways responsiveness after rhinovirus infection is associated with an increase in the number of eosinophils in the sputum.77 Airways hyperresponsiveness in these models may be the result of an increased reflex bronchoconstriction via cholinergic and non-cholinergic excitatory neural pathways. Epithelial necrosis, reduced production of epithelial relaxing factors such as PGE2 and neutral-peptidases, or loosening of tight junctions with overexposure of sensory nerve endings caused by viral replication are the putative underlying mechanisms.78 In animal models infection with parainfluenza 3 virus blocks the ability of β2adrenoceptor agonists to inhibit antigen induced contraction of isolated ASM,79 reduces the activity of muscarinic M2 autoreceptors,80 and increases the number and the releasability of bronchial mast cells.81 The last two effects have been found to be strictly associated with the development of sustained airways hyperresponsiveness. A virus induced overproduction of interferon gamma (IFNγ) from macrophages and lymphocytes has been indicated as the responsible mechanism.82
AIRWAY INFLAMMATION AS A CAUSE OF AIRWAY HYPERRESPONSIVENESS: CONS
Airway inflammation can be observed both in asthmatic and in atopic non-asthmatic subjects.83 ,84 In subjects with allergic rhinitis who have never experienced asthma symptoms both local85 and inhalational86 allergen challenge cause an eosinophilic inflammatory response which cannot be easily distinguished from that seen in asthmatic subjects, but the functional responses are different.86
There is no evidence to suggest that neutrophils play a role in airway hyperresponsiveness in humans.70 In animal models airway neutrophilia induced by sulphur dioxide87 or endotoxin88 is associated with hyporesponsiveness rather than hyperresponsiveness.
The presence of high IgE levels in infancy seems to be a more important predisposing factor for the development of airway hyperresponsiveness than the occurrence of common viral infections in early life.89
Epithelial damage after vaccination with live influenza virus occurs both in healthy and asthmatic subjects but airway hyperresponsiveness develops only in the latter,90 suggesting that individual related factors make an important difference.
The results of studies on the relationships between airway hyperresponsiveness and airway inflammation at baseline are largely inconsistent (table 1). In some instances weak correlations were found between one or more of the inflammatory markers (mainly eosinophils, T lymphocytes and epithelial shedding) and the degree of airway hyperresponsiveness. However, spurious correlations may have resulted from the inclusion of non-asthmatic controls in the regression analysis. Moreover, in an almost equal number of studies no significant correlations were found between airway inflammation and airway hyperresponsiveness. In particular, it was found that airway hyperresponsiveness may be present even in the absence of demonstrable inflammatory cells in the airway lumen or mucosa,114 which suggests that the presence of inflammatory cells in the airways is not necessary to sustain airway hyperresponsiveness.
Studies in human subjects showing (+) and not showing (−) significant correlations between airway inflammation and airway hyperresponsiveness
In lung transplant recipients airway hyperresponsiveness was reported without airway inflammation,115 suggesting that an altered neural control of ASM tone may play a major role in these subjects.
OPEN QUESTIONS
Owing to the lack of evidence that airway hyperresponsiveness and airway inflammation are closely related, there are several questions that must be answered before a causal relationship between airway inflammation and airway hyperresponsiveness in asthma can be established or rejected.
Firstly, can airway hyperresponsiveness develop independent of airway inflammation—for example, as a consequence of inherited abnormalities of ASM contractility or autonomic regulation? This hypothesis cannot be ruled out even if the gaussian distribution of airway hyperresponsiveness in the general population116 and its partial reversibility after pharmacological treatments117or allergen avoidance118 seem to suggest a major role for acquired rather than for inherited factors.
Secondly, is a specific type of inflammation responsible for airway hyperresponsiveness? At present the most appealing hypothesis is that airway hyperresponsiveness is the consequence of repeated episodes of airway inflammation in susceptible subjects. In this connection, allergic inflammation seems to play a major part as atopy with high serum IgE levels is associated with an increased risk of airway hyperresponsiveness in humans.119 In bronchial biopsy specimens of asthmatic subjects the Th2 phenotype has been found to be associated with airway hyperresponsiveness.120 Moreover, in some animal models exposure to the sensitising agent causes an inflammatory response characterised by Th2 lymphocyte phenotype, IL-5 and eosinophil activation,121 which is followed by the development of airway hyperresponsiveness.122 In a murine model, however, airway hyperresponsiveness was obtained even without eosinophil infiltration and was prevented by treatment with anti-IFNγ antibodies, suggesting that eosinophil recruitment and airway responsiveness are differently regulated.123
Thirdly, in which way may airway inflammation lead to the development of airway hyperresponsiveness? One possibility is that the release of mediators (histamine, leukotrienes, PAF, proteases) changes ASM contractility or autonomic regulation. Another possibility is that airway inflammation leads to airway remodelling through the release of chemokines and cytokines. Eotaxin, RANTES, MCP-3, MCP-4, IL-5 and IL-8 promote recruitment, activation and survival of inflammatory cells. IL-4, TGF-β, GM-CSF, TNF and IL-1β modify other aspects of airway biophysiology as antigen processing (Th1–Th2 switch), nitric oxide synthesis, β2 adrenoceptor function and the homeostasis of epithelial cells, fibroblasts, myofibroblasts, and also ASM cells. The former mechanism would be responsible for transient hyperresponsiveness upon exposure to triggers, the latter for sustained baseline hyperresponsiveness.124
Fourthly, are there inherited factors that modulate the consequences of inflammation on airway structure? This possibility is suggested by epidemiological data125 indicating that a child with an allergic parent has an increased risk of developing the same allergic disease as affects the parent, whereas the risk of developing a different allergic disease is not significantly different from that for the general population.
Conclusions
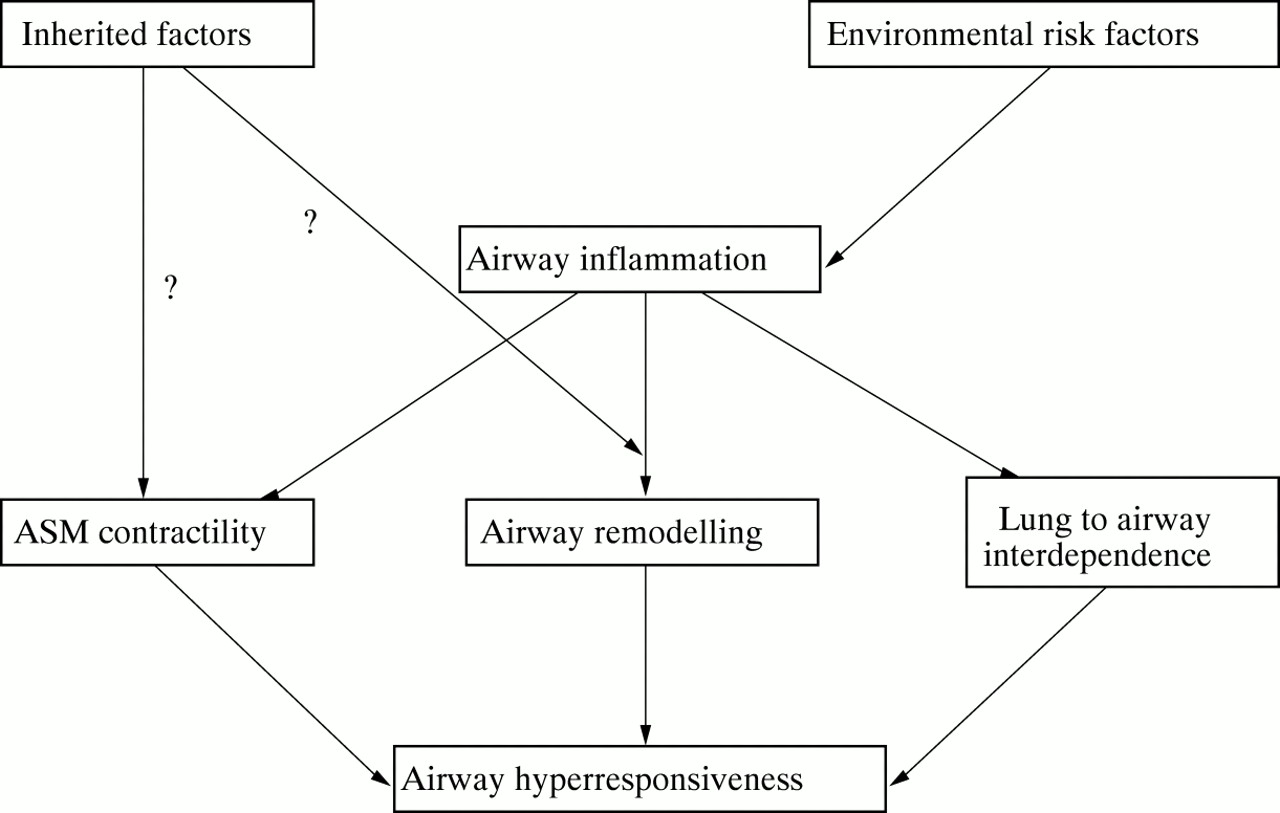
Airway inflammation and airway hyperresponsiveness, two major characteristics of bronchial asthma, are loosely related to each other. It seems that the presence of inflammatory cells in the airways is neither sufficient nor necessary for the development of airway hyperresponsiveness. This would imply that an altered response of the target organ is a prerequisite for airway hyperresponsiveness to develop. In this scenario, chronic airway inflammation is likely to play a key role as a stimulus for structural changes (airway wall remodelling, changes in airway to lung interdependence, changes in ASM contractility) affecting the organ response to acute stimuli (fig 1) . A practical conclusion is that no inferences about airway hyperresponsiveness can be made from measurements of airway inflammation and vice versa.

{kind=link}
Hypothetical mechanisms and pathways of airway hyper-responsiveness.