Article Text

Statistics from Altmetric.com
Renal and hormonal abnormalities, usually manifested as oedema or hyponatraemia, are encountered frequently in patients with chronic obstructive pulmonary disease (COPD). The exact incidence of clinically significant oedema and hyponatraemia has not been documented. In advanced disease some degree of oedema is observed in a large proportion of patients; the pattern of hyponatraemia parallels that of oedema, but with a lower frequency. In the past, oedema in patients with COPD has been attributed to “cor pulmonale with backward heart failure”—that is, pulmonary hypertension induced by hypoxia and by structural changes in pulmonary arteries, increased systemic venous pressure, and reduced cardiac output. The onset of oedema is a poor prognostic factor; Renzetti and coworkers1 reported a four year mortality rate of 73% in patients with cor pulmonale compared with 53% for the whole group. Whether this reflects the advanced stage of the disease, the indirect effect of chronic diuretic therapy, or some undetermined insult on the function of critical organs is unclear. What is clear is that oedema formation in COPD is not cardiac in origin: in most patients, even when they are frankly oedematous, cardiac output is adequate for the body’s metabolic demands2 ,3 unless there is significant co-existent cardiac disease.
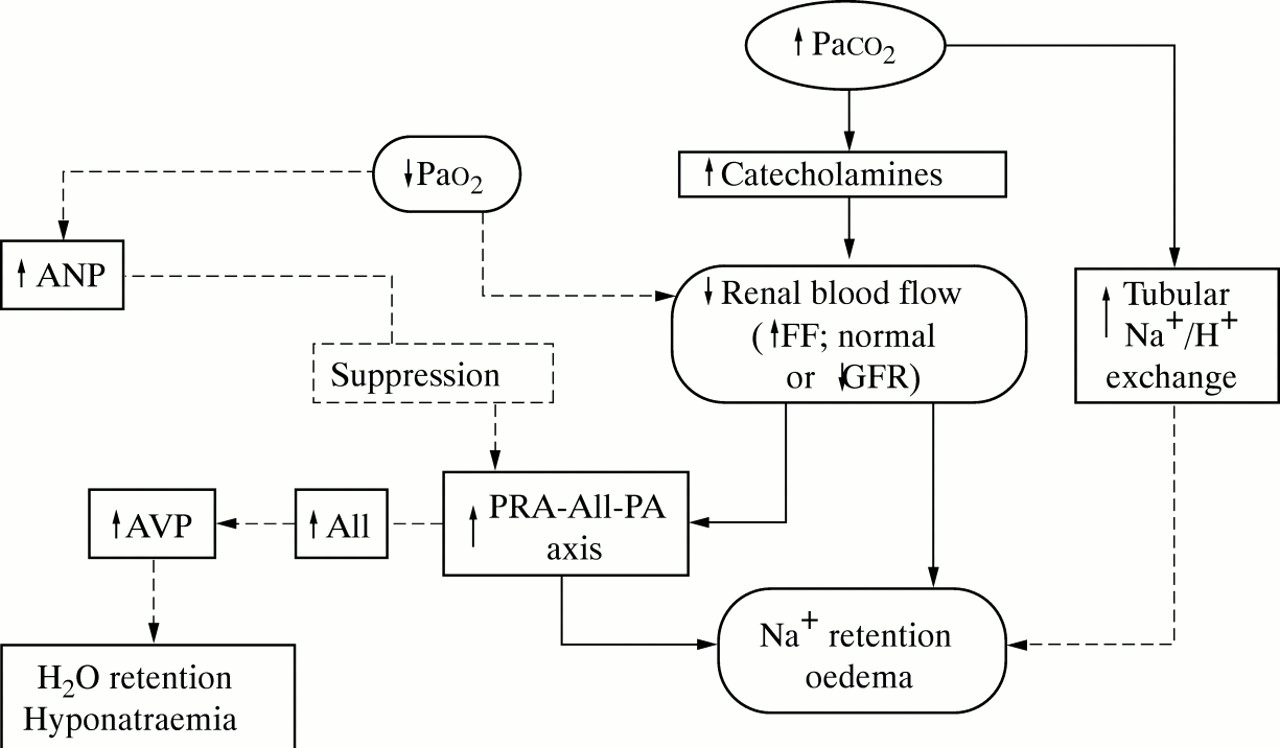
In 1960 Campbell and Short4 pointed out that, in patients with COPD, oedema is almost invariably associated with carbon dioxide (CO2) retention. They concluded that, in hypoxaemic normocapnic patients with chronic diffuse lung disease such as pulmonary fibrosis, oedema is uncommon and, in this setting, transient worsening in blood gas tensions during exercise or sleep, for example, should be suspected and ruled out. Since then sodium (Na+) retention in COPD has been considered to be the result of electrochemical imbalance (enhanced renal tubular H+/Na+ exchange with attendant increase in Na+ reabsorption) and/or renal haemodynamic abnormalities (hypercapnia/hypoxaemia-mediated reduction in effective renal plasma flow (ERPF), increased filtration fraction (FF) and consequent increase of peritubular oncotic pressure, a recognised cause of Na+retention).5 More recently experimental evidence has accumulated in support of the hypothesis that, in the advanced stages of COPD, imbalances in hormones that regulate body Na+ and water homeostasis—namely, the renin-angiotensin-aldosterone axis and the arginine-vasopressin system—are potential contributors to oedema and hyponatraemia.6 Table 1 summarises the abnormalities of arterial blood gases, renal and hormonal indices during the progression of the disease. The focus of this review is to highlight the current knowledge (fig 1) on renal/hormonal function disturbances in COPD and, briefly, their therapeutic implications.
Renal and hormonal abnormalities in COPD during the progression of the disease
{kind=link}
Renal and hormonal abnormalities induced by chronic hypercapnia and possibly aggravated by severe hypoxaemia in the pathogenesis of sodium and water retention in COPD. Solid lines indicate established mechanisms; broken lines indicate non-proven pathways. ANP = atrial natriuretic peptide; AVP = arginine vasopressin; GFR = glomerular filtration rate; PRA = plasma renin activity; AII = angiotensin II; PA = plasma aldosterone; FF = filtration fraction; Pao 2, Paco 2 = arterial oxygen and carbon dioxide tensions. Modified from Farber and Manfredi.6
Renal abnormalities
The most consistent alteration in renal function in hypoxaemic hypercapnic patients with COPD is the reduction in ERPF.7-9 In patients with moderate hypoxaemia, mild hypercapnia, normal cardiac output, and no evidence of intrinsic renal disease, a significant reduction in ERPF associated with normal glomerular filtration rate (GFR) and increased FF was reported as long ago as 1961.10 By contrast, the GFR is usually preserved until the very late phases of the disease. The reduction in ERPF in the presence of a normal GFR increases FF and consequently Na+retention. These findings indicate that, under the conditions investigated, arteriolar renal resistances are increased, perhaps because of local adrenergic discharge secondary to hypercapnia. In the initial phase of COPD renal perfusion is usually normal but, as the disease worsens, particularly as CO2 retention develops, renal blood flow decreases.11 ,12Paco 2 has been found to correlate inversely with ERPF and with the ability to excrete Na+ and water; in some oedematous patients a reduction in renal flow of as much as 63% has been reported.13 Hypercapnia may cause renal vasoconstriction directly9 and indirectly by stimulating sympathetic tone as reflected by the increase in the circulating levels of norepinephrine.14 ,15 Greater sympathetic tone enhances tubular Na+ reabsorption by reducing ERPF and/or redistributing renal blood flow16. In stable hypercapnic COPD patients total body Na+ is increased, with or without clinical evidence of oedema.17 Hypoxaemia alone does not seem to induce significant changes in renal haemodynamics and/or in Na+ and water homeostasis, although oxygen administration has been shown to exert a vasodilator effect—for example, increased renal arterial velocity—in hypoxaemic normocapnic COPD patients.18 In hypercapnic patients neither short term oxygen therapy nor infusion of low dose dopamine improve renal haemodynamics19; however, correction of hypoxaemia by long term oxygen therapy in hypercapnic patients with chronic respiratory failure may result in significant natriuresis,20 due possibly to amelioration in lung mechanics with a reduction in Paco 2(bronchodilator effect with lower airways resistances) or to a reduction in the levels of plasma renin activity (PRA) and plasma aldosterone (PA).21
Hormonal abnormalities
As previously mentioned, plasma catecholamine levels increase in response to CO2 retention early in the disease, an index of increased renal sympathetic tone, leading to Na+ retention. Late in the course of the disease the ability to excrete Na+ and water is further aggravated by activation of the PRA-PA system,22-24 a state of “secondary hyperaldosteronism”. In many oedematous patients in whom low ERPF values and mild reduction in GFR can be demonstrated, high circulating levels of PRA, angiotensin II, and PA are usually observed. Patients with COPD may have levels 2–3 times higher than normal subjects but usually lower than the levels observed in patients with congestive heart failure.25 In animal studies acute hypoxia has been reported to induce an increase in PRA26 ,27 whereas chronic hypoxia produces variable results.28 ,29 In COPD the PRA-PA axis is stimulated only when GFR starts to fall as a result of a significant reduction in ERPF.22 Hypoxaemia alone does not stimulate the PRA-PA axis: correction of hypoxaemia has been reported to reduce PRA and PA values in one study21 but different results were obtained in another study by the same investigators.20
As a result of the stimulation of the PRA-PA axis, the activity of the angiotensin converting enzyme (ACE) is increased and high values of angiotensin II may be observed. The results of studies on the effect of ACE inhibitors are controversial. In a group of patients with COPD captopril induced a significant increase in Na+ excretion without any concomitant changes in ERPF or PA.30 The opposite result was observed in a study reported by Stewart et al 31 in which, despite the observed concomitant reduction in PA, Na+ excretion was not influenced by the administration of perindopril.
Significant hyponatraemia is present in a considerable number of oedematous COPD patients. In these patients the levels of arginine vasopressin (AVP), an antidiuretic hormone, are inappropriately high for the level of plasma osmolality.21 ,22 ,32 The mechanism that underlies this abnormality is not fully understood. In normal subjects AVP release is mainly controlled by plasma osmolality. In patients with COPD a non-osmotic mechanism should be invoked since increased AVP is inappropriate for the plasma osmolality. Although hotly debated,33 experimental data exist to suggest that an increase in angiotensin II may stimulate AVP directly. Stimulation of baroceptors in oedematous patients with low circulating blood volume has also been suggested.34 Not all patients with high AVP levels have hyponatraemia; in some patients Na+ levels may remain normal until excessive water intake occurs.
As in patients with chronic heart failure, increased levels of atrial natriuretic peptide (ANP) have been reported in oedematous COPD patients.25 ,34 In addition, ANP levels have been shown to correlate inversely with Pao 2.34What is not completely understood is why high circulating ANP levels in oedematous patients with COPD do not promote Na+ and water excretion. In normal subjects endogenous ANP release in response to acutely increased atrial pressure35 or infusion of exogenous ANP36 results in prompt natriuresis and diuresis. An acute increase in ANP induces a suppression of the PRA-PA axis in normal subjects.37 ,38 Experimental data in COPD have shown that the PRA-PA axis is not suppressed by an increase in ANP induced by potent stimuli such as exercise39 or application of lower body positive pressure.25 Thus, the PRA-PA and ANP systems appear to be dissociated, a condition similar to that observed in normal subjects during exercise.40 It is logical to assume that, in oedematous patients with COPD, the effect of reduced ERPF on PRA-PA release is more potent than the effect of ANP on PRA-PA suppression. ANP therefore seems to be capable of correcting acute central volume but in chronic oedematous states it has little influence on Na+ overload.
In view of the above findings, it is clear that the therapeutic intervention required to control oedema in advanced COPD is quite different from that used in congestive heart failure. A correct therapeutic strategy should include the reversal of hypoxaemia and the improvement of lung mechanics by reducing bronchial secretions and promoting maximal brochodilatation. The role of non-invasive mechanical ventilation—which may be used to reduce excessive CO2—has not been investigated. Hyponatraemia, when present, should be treated with water restriction that usually results in either stabilisation or a slight increase in the plasma sodium concentration. The effect of ACE inhibitors is still debated. The use of digitalis should be avoided unless intrinsic heart disease with a low output is documented; in the presence of severe hypoxaemia this drug may expose patients to an increased risk of arrhythmias. Although some investigators have reported a favourable response to the administration of diuretics,41 these drugs should be used with caution since they may result in hypochloraemic metabolic alkalosis which may lead to hypoventilation with worsening of blood gas tensions and further stimulation of plasma renin activity.
In summary, in the last few years the pathogenesis of Na+and water retention in COPD has been revised. It is now clear that renal and hormonal abnormalities, induced by hypercapnia and possibly aggravated by hypoxaemia, play a pivotal role in the development of oedema and hyponatraemia. The revisions have led to a new therapeutic approach to the oedematous COPD patient.