Article Text

Abstract
BACKGROUND It has been reported that pranlukast reduces the antigen induced immediate and late phase asthmatic responses, airway hyperreactivity to acetylcholine, and pulmonary eosinophil accumulation in guinea pigs. A study was undertaken to test the hypothesis that pranlukast may reduce the number of inflammatory cells in the bronchial mucosa of patients with asthma.
METHODS A double blind, placebo controlled study was performed in 17 mild to moderate asthmatic subjects to examine changes in inflammatory cell infiltration in response to pranlukast (225 mg orally twice per day for four weeks). Comparisons of the mean daily β2 agonist use, symptom score, FEV1 percentage predicted, and airway methacholine responsiveness were made before and after treatment. Using fibreoptic bronchoscopy, bronchial biopsy specimens were obtained before and after treatment with either pranlukast (n = 10) or placebo (n = 7). Immunohistology was performed using monoclonal antibodies for CD3, CD4, CD8, CD68, NP57, AA1, EG1, EG2, γGTP and CD19.
RESULTS When the pranlukast and placebo treated groups were compared there were decreases in β2agonist use, symptom score, and airway methacholine responsiveness after pranlukast but no increase in FEV1 was seen. The clinical response in patients treated with pranlukast was accompanied by a reduction in CD3 (median difference –37, 95% confidence interval (CI) –69 to –1; p<0.05), CD4 (median difference –28, 95% CI –49 to –8; p<0.01), AA1 (median difference –15, 95% CI –26 to 0; p<0.05) and EG2 positive cells (95% CI –35 to 0; p<0.05), but not in EG1 positive eosinophils, γGTP positive cells, and CD19 positive plasma cells.
CONCLUSIONS These results support the view that pranlukast may act by inhibition of bronchial inflammation in patients with asthma.
- leukotriene receptor antagonist
- asthma
- pranlukast
Statistics from Altmetric.com
Bronchial asthma is a chronic inflammatory disease associated with infiltration of eosinophils, mast cells, and T lymphocytes into the airway wall.1 Cysteinyl leukotrienes (LTC4, LTD4 and LTE4), which are released from inflammatory cells, are potent bronchoconstrictor agents,2 ,3 increase vascular permeability, and may contribute towards the development of mucosal oedema4 ,5 and excessive mucus secretion.6Furthermore, increased levels of leukotrienes have been reported in the sputum, urine, bronchoalveolar lavage fluid, plasma, and nasal secretions of asthmatic patients.7-11 As such, therapeutic strategies designed to limit the production and/or release of these mediators have attracted considerable attention in the treatment of bronchial asthma.
In vitro studies have shown that pranlukast antagonises the action of LTC4, LTD4 and LTE4 within the airway.12 We hypothesised that the clinical action of pranlukast is a result of its ability to reduce the inflammatory cells within the airways of asthmatic subjects. We have therefore undertaken a double blind placebo controlled trial in patients with atopic asthma to assess the effect of pranlukast on immunocytochemical markers of airways inflammation. To delineate the action of this drug on leukotriene metabolism we also examined the ability of pranlukast to activate γGTP. In addition, we have correlated its effect on the airway mucosa with changes in airways responsiveness to methacholine and forced expiratory volume in one second (FEV1).
Methods
PATIENTS
Twenty one subjects were recruited from Toho University Hospital. All fulfilled the American Thoracic Society (ATS) criteria for asthma,13 had typical clinical symptoms, documented airways reversibility, and increased airways responsiveness to methacholine. The study was approved by the ethics committee of our institute and informed consent was obtained from all patients before entry into this study.
None of the patients had received inhaled or oral corticosteroids, or any other anti-inflammatory medication such as disodium cromoglycate, nedocromil sodium, or theophylline within the previous four months. Ten of the subjects took intermittent β2 agonists only when necessary. All patients were atopic as defined by a positive skin prick test to one or more common aeroallergens. Additional antigen inhalation tests were carried out with antigens to ensure their atopic status. The antigen dilutions were inhaled by a De Vilbiss 646 nebuliser (De Vilbiss Co, Somerset, Pennsylvania, USA) at a constant flow rate of 5 l/min. FEV1 was measured 10 minutes after administration of saline for two minutes to obtain the baseline value. For each of the antigens FEV1 was measured in the same way in ascending order of concentration and repeated 20, 30, 60 and 120 minutes after inhalation of the highest concentration of each antigen. Reduction in FEV1 exceeded 20% in every patient.
All of the subjects were lifelong non smokers and none had any other serious lung disease as assessed by a chest radiograph obtained within one month before entry to the study. Patients with FEV1 of <50% predicted were excluded.
STUDY DESIGN
The study was of a randomised parallel group design. Before entry the patients were instructed in the performance of spirometric tests according to American Thoracic Society standards. Following a two week observation period fibreoptic bronchoscopy was performed. Patients were randomised to receive either pranlukast (225 mg twice a day) or matched placebo for four weeks. All doses of study medication were administered with water within two hours after a meal. No drugs other than inhaled β2 agonists as required were allowed during the trial. Patients were instructed to make a daily record of their use of inhaled β2 agonists. During the observation and treatment periods the patients recorded the severity of cough, expectoration of sputum, disturbance in daily activities and sleep due to asthma, and the consumption of the given drug in their patient diary. Bronchoscopic examination was performed twice: at the end of the observation period and at the end of the four week period of treatment. One week before the bronchoscopic examination lung function tests and airway hypersensitivity were measured.
AIRWAYS RESPONSIVENESS TO METHACHOLINE
The use of any bronchodilator was discontinued for 12 or more hours before the methacholine induced airway hypersensitivity was measured by the method of Takishima et al 14using an Astograph (Chest Co, Tokyo, Japan) direct writing recorder which gives dose-response curves of respiratory resistance (Rrs) during continuous inhalation of methacholine in isotonic saline in stepwise incremental concentrations (49, 98, 195, 390, 781, 1563, 3125, 6250, 12 500, and 25 000 μg/ml). Saline solution was inhaled first, followed after one minute by successive inhalations of increased concentration of methacholine. The minimum dose of methacholine (Dmin) was used as the indicator of bronchial sensitivity—that is, the amount of the cumulative dose at the point where the reciprocal of the Rrs decreased linearly. Dmin was scaled by a unit equal to one minute of a 1.0 mg/ml aerosol inhalation of methacholine.
SCORING OF SYMPTOMS
Six symptom scores were recorded daily on the diary card: (1) wheeze and (2) cough during the day using a graded scale as none = 0, mild = 1, moderate = 2, severe = 3; (3) amount of sputum graded as small = 1, large = 2; (4) limitation of daily activity graded as none = 0, mild = 1, moderate = 2, severe = 3; (5) nocturnal wheeze and (6) nocturnal cough graded as none = 0, mild = 1, moderate = 2, severe = 3. The results were expressed as the daily mean of the sums of the six scores. The mean initial symptom score was calculated from the scores recorded for the two week observation period. In the same way the mean symptom score after treatment was calculated from those recorded during weeks 3 and 4 of treatment.
BIOPSY OF BRONCHIAL MUCOSA
Biopsy specimens were obtained during a bronchoscopy performed according to published guidelines. The patients were premedicated with atropine sulphate (0.6 mg im) and diazepam (5 mg iv) and two puffs (200 μg) of salbutamol were then given using 4% lignocaine for local anaesthesia of the pharynx and the larynx. The bronchoscope (BF-20, Olympus Co, Tokyo, Japan) was inserted via the mouth following surface anaesthesia of the trachea and the bronchi with 2% lignocaine. Oxygen was administered via a nasal cannula at 4 l/min and the arterial oxygen saturation was monitored with a pulse oximeter (Biox 3740, Pulse Oximeter, Ohmeda, Louisville, Kentucky, USA). Using alligator forceps (FB-15C, Olympus) three bronchial biopsy specimens were obtained at the right main bronchus, the bifurcation of the right middle lobe; and the right B6 inlet. At the second bronchoscopic examination tissue samples were taken from the same site as on the first occasion.
HISTOPATHOLOGICAL EXAMINATION
The tissue specimens were covered in ornithine carbamyl transferase (OCT) compound, rapidly frozen in dry ice-acetone, and stored at –70°C until further use. Specimens from all the tissues were stained with haematoxylin and eosin. Cryostat sections (4 μm) were fixed for 15 minutes in cold acetone, washed five times with phosphate buffered saline (PBS) for five minutes, and then treated for 30 minutes with 10% normal porcine serum. The following monoclonal antibodies were then added and allowed to react for 60 minutes: CD3 (T cells), CD4 (“helper” T cells), and CD8 (suppressor T cells; Becton Dickinson, UK), CD68 (macrophages), NP57 (neutrophil elastase), AA1 (mast cell tryptase; Dako Ltd, UK), EG1 (Nichirei Ltd, Japan) and EG2 (Kabi Pharmacia, Uppsala, Sweden) which stain eosinophil for the cleaved form of eosinophilic cationic protein. Antibodies to γGTP (Cosmobio Ltd, Japan), an enzyme which metabolises leukotrienes, and CD19 (plasma cells; Becton Dickinson, UK) were also used. After washing with PBS, peroxidase-labelled anti-mouse immunoglobulin G (IgG) was added for one hour at room temperature, washed, and treated with NaN3 (65 mg/dl) to prevent non-specific binding due to endogenous peroxidase in the eosinophils and neutrophils. The reaction was developed with 3,3′-diaminobenzidine (DAB). Sections were counterstained with methylene green before examination. For negative control the primary antibody was replaced by either non-specific rabbit immunoglobulin or PBS.
COUNTING OF POSITIVELY STAINED CELLS
The specimens were all coded and examined blind using a wide field microscope (BH2, Olympus) at a magnification of ×400. The observer counted the number of cells of the whole specimen. The stained cells were counted only in intact mucosa, excluding epithelium, mucosa, muscle and vessels. The contour of the tissue in which cells were counted was traced with computer software (NEC Co., Tokyo, Japan) on a video display (ITC-370, Olympus) and the area was calculated to determine the number of cells per unit area (mm2) tissue. The within observer χ2 test for goodness of fit for five repeated counts (df = 4) was less than 5% for all the monoclonal antibodies studied.
STATISTICAL ANALYSIS
The mean symptom scores were calculated from the daily mean of the sum of the six symptoms both for the two weeks of the baseline period and for the last two weeks of the treatment period, during which time β2 agonist use was also measured. The clinical response and cell counts during pranlukast treatment were compared with those during placebo using the Mann-Whitney U test on the delta values (difference before and after treatment) for the two treatment groups. Results were expressed as the estimated median difference between delta values with 95% confidence intervals. The Wilcoxon matched pairs signed rank test was used to examine changes within groups. p values of less than 0.05 were considered statistically significant.
Results
CLINICAL RESPONSE
Seventeen patients completed the study. Two patients (one from the pranlukast group and one from the placebo group) discontinued for personal reasons and two patients (both from the placebo group) discontinued because of an upper respiratory tract infection. There were no reported side effects during treatment. There were no significant differences in age, sex, total IgE, FEV1 (% predicted), and airway methacholine responsiveness between patients receiving pranlukast or placebo at baseline (table 1). The serum concentration of pranlukast ranged from 100 to 736 ng /ml. Compared with placebo, decreases in β2 agonist use and asthma symptom scores were observed with pranlukast treatment (p <0.01; table2). No significant improvement in FEV1 was observed in patients treated with pranlukast (table 2, fig 1). Pranlukast treatment was accompanied by a decrease in airway responsiveness which was significant when compared with the change following placebo treatment (p <0.05; table 2, fig 1).
Clinical data of patients
Change in patient reported end points
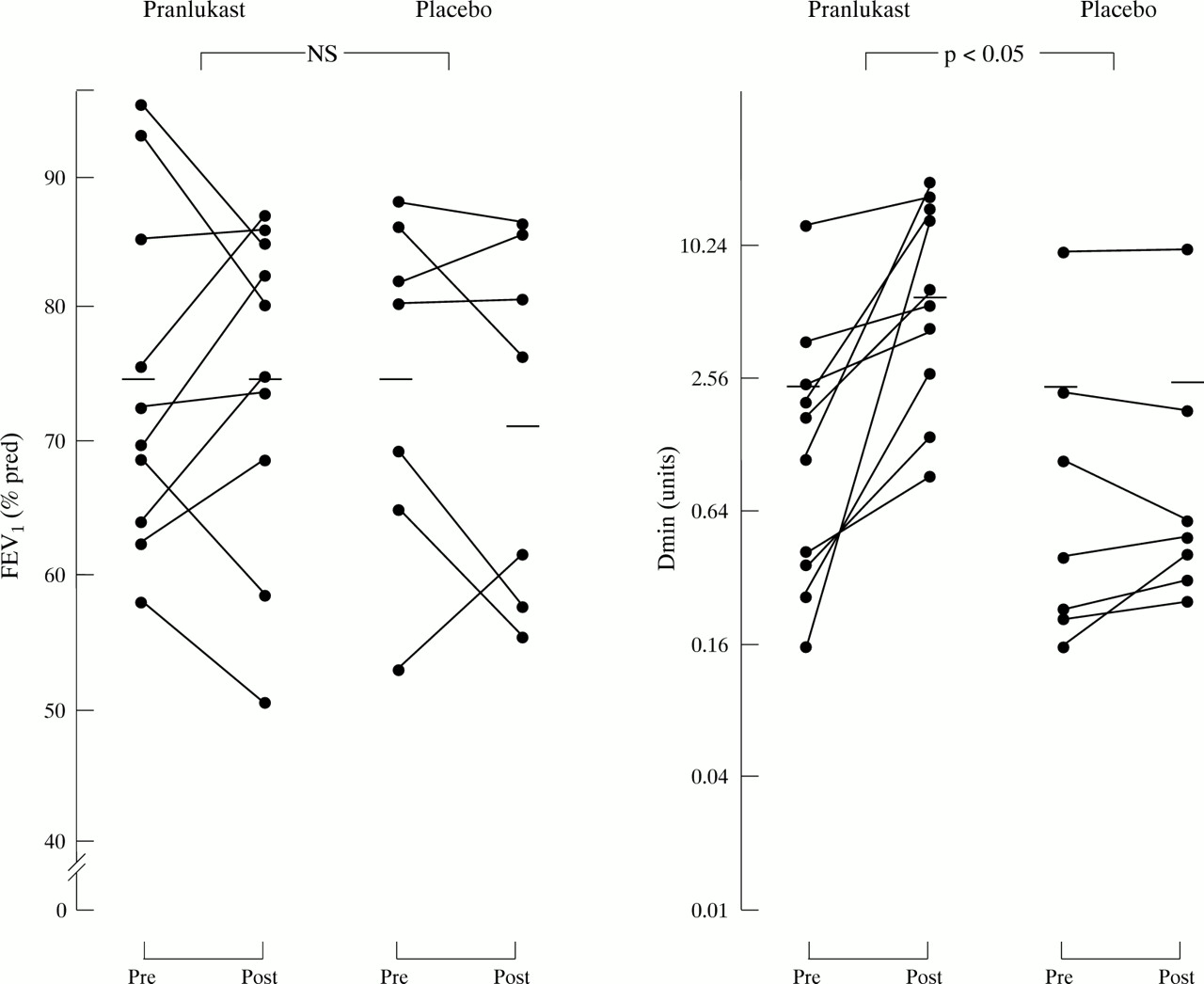
Percentage predicted forced expiratory volume in one second (FEV1) and minimum dose of methacholine as a measure of bronchial responsiveness before and after treatment with pranlukast or placebo. Horizontal bars are mean values.
HISTOPATHOLOGICAL RESULTS
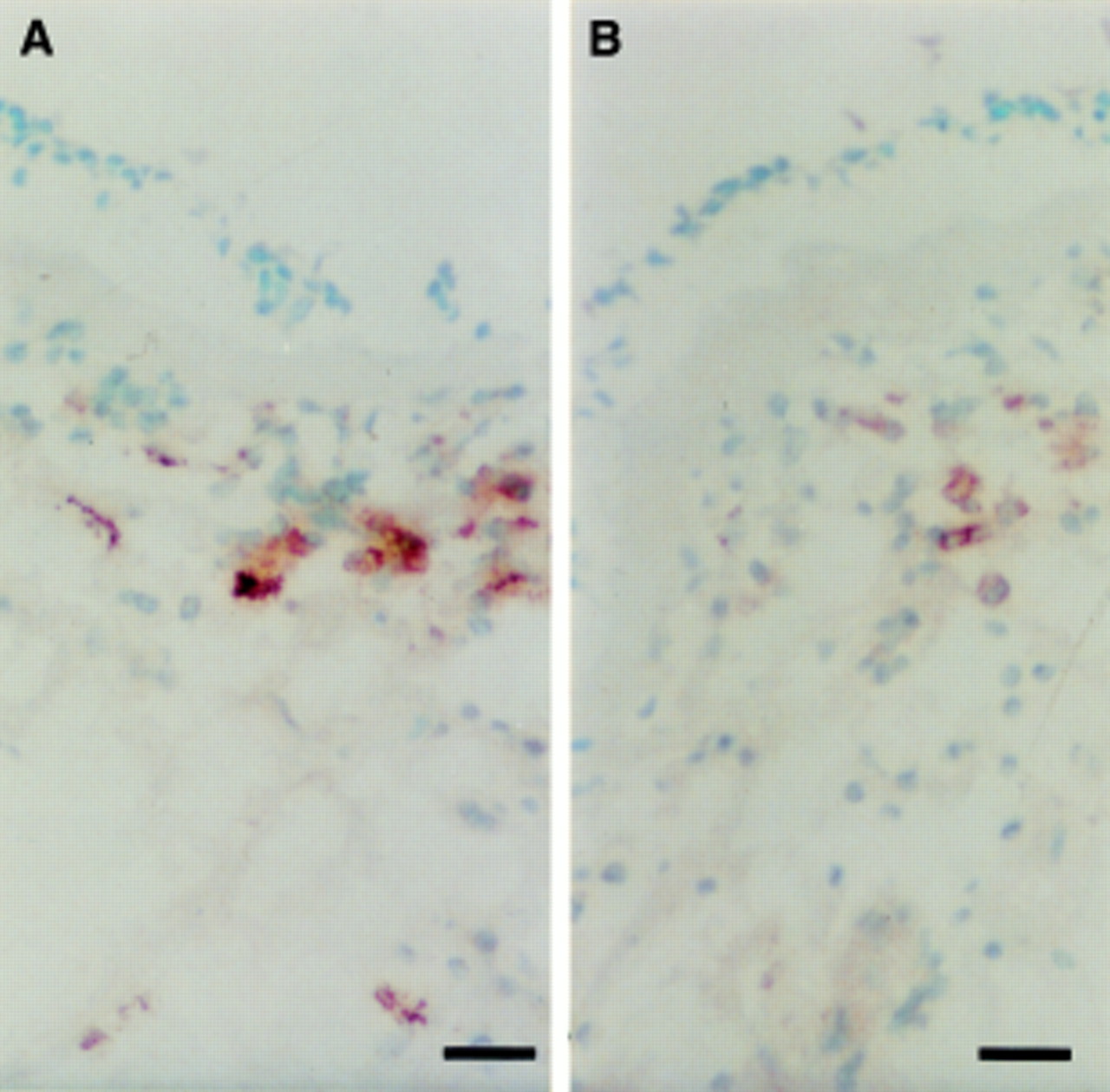
At baseline there were no significant differences between subjects receiving pranlukast and those receiving placebo in bronchial mucosal immunohistochemical tests for activation markers (table 3). Significant reductions in CD3, CD4, AA1, and EG2 positive cells were seen following treatment with pranlukast (figs 2 and 3), and there was also a significant reduction in the numbers of CD3, CD4, AA1 and EG2 positive cells compared with the placebo treated group. In contrast, changes in CD8 positive cells, CD68 (macrophage), NP57 (neutrophils), EG1, γGTP, and CD19 (plasma cells) within the pranlukast treated group were not significant and between group comparisons did not show significant differences. No significant changes were observed in cell numbers in biopsy specimens from the placebo treated group. Figure 4A and B illustrate the changes in the numbers of EG2 positive eosinophils from a single patient.
Number of cells per mm2 before and after treatment with pranlukast and placebo
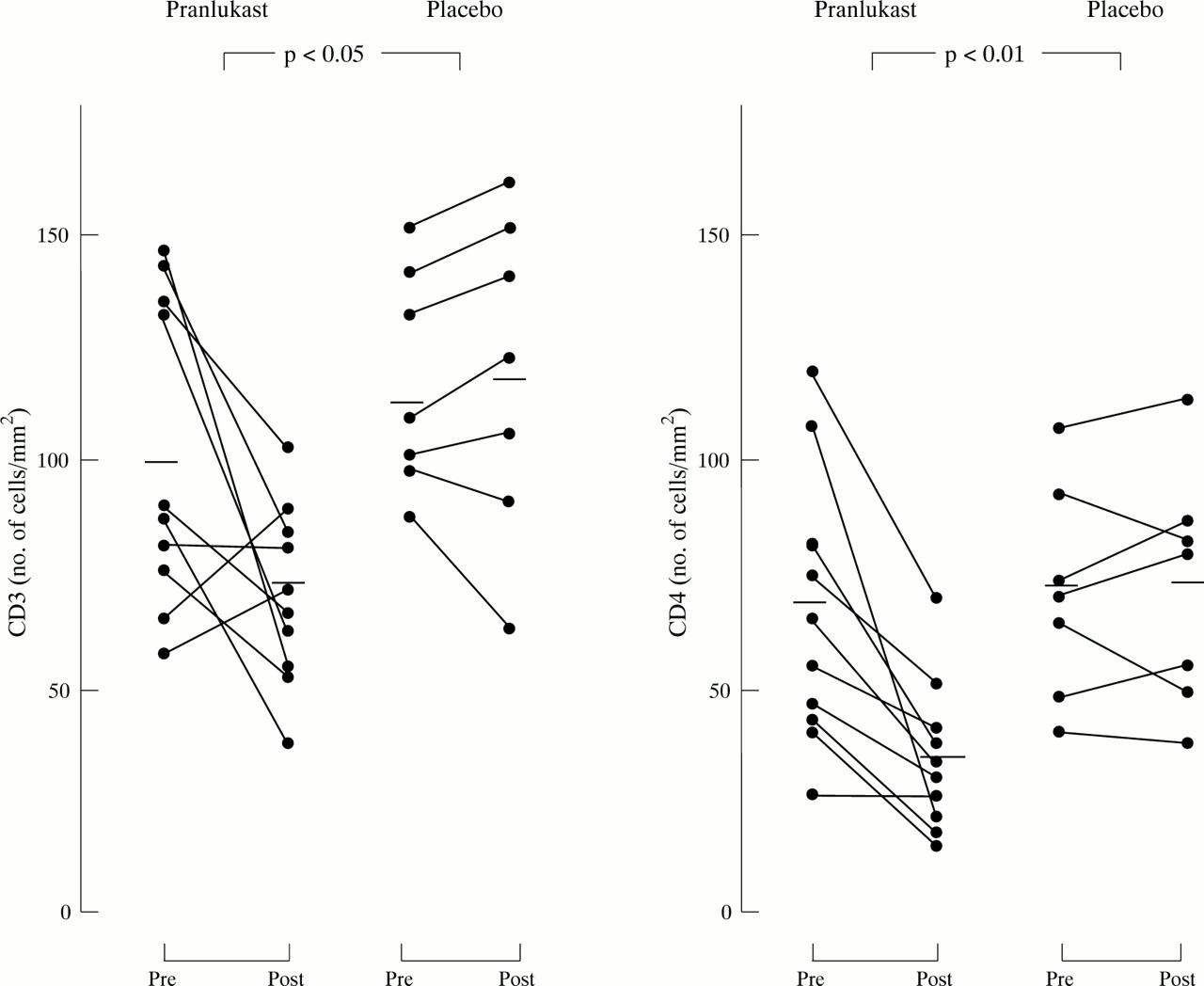
Individual T lymphocyte (CD3 and CD4) counts in the bronchial mucosa expressed as numbers of positive cells per square millimeter before and after treatment with pranlukast or placebo. Horizontal bars are mean values.

Individual mast cell (AA1) and eosinophil (EG2) counts in the bronchial mucosa expressed as numbers of positive cells per square millimeter before and after treatment with pranlukast or placebo. Horizontal bars are mean values.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Biopsy specimen of bronchial mucosa stained for EG2 positive eosinophils (A) before and (B) after treatment. Bar = 20 μm.
Discussion
In this double blind, placebo controlled study we have shown that decreased airways responsiveness in asthma after pranlukast treatment was accompanied by a decrease in the number of bronchial inflammatory cells in the airways. Pranlukast improves objective measurements of asthma compared with placebo, causing significant improvements in asthma symptom scores and a decrease in as needed β2agonist use.
An LTD4 antagonist, zafirlukast, has been shown to attenuate allergen induced bronchoconstriction and hyperreactivity in atopic asthmatic subjects.15 Selective antagonists of LTD4 and LTE4 only partially inhibit the antigen induced contraction of guinea pig trachea.16 ,17 In contrast, pranlukast has been found to possess antagonist activity against LTC4 as well as LTD4 and LTE4.12 Pranlukast also reduces antigen induced bronchoconstriction, airway hyperreactivity to acetylcholine, and pulmonary eosinophil accumulation in guinea pigs.18 The mean value of Dmin after the administration of pranlukast was significantly greater than the value after placebo administration, but the values of FEV1 were not altered by pranlukast. The mechanism by which the cysteinyl leukotrienes affect the human airway is not completely understood. However, the clinical improvements seen in patients receiving multiple dose pranlukast provide evidence to suggest that these leukotrienes act as mediators for asthma symptoms. In clinical trials of other leukotriene inhibitors treatment with zafirlukast and montelukast resulted in significant improvements in pulmonary function tests and decreases in asthma symptoms and use of β agonists.19 ,20 In a trial of zileuton (a 5-lipoxygenase inhibitor) patients receiving a dose of 2.4 mg/day experienced significant improvements in airway function and hyperresponsiveness.21 This study indicated that pranlukast did not improve pulmonary function in asthmatic patients but it did improve bronchial hyperresponsiveness. Fujimura et al 22 suggested that subthreshold concentrations of leukotrienes in the bronchial tissues, which have no effect on bronchomotor tone per se, may be involved in the development of bronchial hyperresponsiveness in asthma. The magnitude and precise mechanism of this effect on airway function and hyperresponsiveness require the investigation of long term clinical benefits of pranlukast.
It has been reported that cysteinyl leukotrienes induce eosinophilia in guinea pig airways and that the LTD4 receptor antagonist, MK- 571, significantly inhibits LTD4 and antigen induced eosinophil migration.23 However, no studies have examined the anti-inflammatory effects of pranlukast on the bronchial mucosal cell infiltrate in asthma. In this placebo controlled study we observed a significant decrease in the number of EG2 positive eosinophils, AA1 positive mast cells, and CD3 and CD4 positive T cells. We are uncertain of the significance of the observed decrease in these inflammatory cells after treatment with LTC4, LTD4 and LTE4 receptor antagonist. It seems reasonable to speculate that, by stabilising eosinophils, pranlukast inhibits the release of some cytokines, thereby minimising eosinophil infiltration. Our data support this hypothesis as there was a significant reduction in activated eosinophils but no difference in EG1 positive eosinophils (or inactivated eosinophils). Most chemokines released from eosinophils with two N-terminal conserved cysteine residues in juxtaposition stimulate monocyte cytotoxic activity and upregulate adhesion molecules. They also augment T cell proliferation and cytokine production. We therefore assume that treatment with pranlukast inhibits the activation of eosinophils and the influx of inflammatory cells into the airways. The present study suggests that part of the beneficial effect of pranlukast might be related to its apparent ability to reduce the number of local inflammatory cells. Further studies on bronchoalveolar fluid concentrations of chemokines and leukotrienes after administration of pranlukast will be required.
Taniguchi and co-workers showed that treatment with pranlukast for one week did not alter percentage changes in respiratory resistance 10 minutes after challenge. However, they also showed that the treatment significantly reduced percentage maximum changes in respiratory resistance, as well as percentage changes in these two indices 20–60 minutes after the exposure compared with placebo treatment.24 It is also reported that the maximal response occurred 4–16 minutes after inhalation of LTD4. In contrast, the maximal response was delayed and occurred 10–20 minutes after inhalation of LTC4. This is similar to the time required for significant amounts of bioconversion of LTC4 to LTD4 by γGTP to occur in vitro.25 From these results we speculated that the effect of pranlukast may also be due to the inactivation of γGTP, resulting in the suppression of the production of LTD4 and taking 20 minutes to change respiratory resistance. However, our results suggest that treatment with pranlukast did not reduce the existing γGTP positive cells. The inhibitory activities of pranlukast may not be mediated by the blockade of γGTP activity but might occur through the direct interaction with LTC4, LTD4 and LTE4 receptors. Alternatively, it is possible that the power of this study may not have been sufficient to detect differences in γGTP positive cells.
With bronchial biopsy specimens we have been shown that pranlukast not only provided benefit in the treatment of asthma but also affected the number of different inflammatory cells.
Acknowledgments
We are indebted to Omar Ghaffar, Kuniharu Ono, Eleanor Minshall and Qutayba Hamid for editorial assistance.