Article Text

Statistics from Altmetric.com
Emphysema is a chronic progressive lung disease characterised by abnormal permanent enlargement of airspaces as a result of destruction of alveolar walls.1 Most patients develop emphysema as a consequence of smoking but 1–2% of patients with emphysema develop the condition as a result of a genetic deficiency of the plasma proteinase inhibitor α1-antitrypsin. The two common deficiency variants of α1-antitrypsin, S and Z, result from point mutations in the α1-antitrypsin gene2-4 and are named on the basis of their slower electrophoretic mobility on isoelectric focusing analysis compared with the normal M allele.5 S α1-antitrypsin (264Glu→Val) is found in up to 28% of Southern Europeans and, although it results in plasma α1-antitrypsin levels that are 60% of the M allele, it is not associated with any pulmonary sequelae. The Z variant (342Glu→Lys) results in a more severe deficiency that is characterised, in the homozygote, by plasma α1-antitrypsin levels of 10% of the normal M allele and by levels of 60% in the MZ heterozygote (50% from the M allele and 10% from the Z allele). The Z mutation results in the accumulation of α1-antitrypsin in the rough endoplasmic reticulum of the liver (fig 1A) and predisposes the homozygote to juvenile hepatitis, cirrhosis,6 and hepatocellular carcinoma.7 Z α1-antitrypsin inclusions are associated with abnormal liver function tests in over 90% of Z homozygotes in the first year of life but only 10–15% of these develop the prolonged cholestatic jaundice that can progress to cirrhosis and the requirement for hepatic transplantation.6 ,8

(A) Electron micrograph of a hepatocyte from the liver of a child with Z α1-antitrypsin deficiency. The arrow shows the accumulation of α1-antitrypsin within the rough endoplasmic reticulum and the distortion of the hepatocellular architecture that predisposes to cell death and cirrhosis (reproduced from ref 26 with permission). (B) High resolution CT scan from a patient with Z α1-antitrypsin deficiency showing the characteristic basal panlobular emphysema rather than the apical centrilobular disease seen in smokers who have normal levels of α1-antitrypsin.
The role of α1-antitrypsin is to protect the tissues against enzymatic digestion by neutrophil elastase.9 The low circulating levels are unable to inhibit this proteinase and predispose the Z homozygote to early onset panlobular emphysema,10 bronchiectasis,11 and vasculitis.12 Alpha1-antitrypsin deficiency related emphysema is predominantly panlobular and basal compared with the centrilobular upper lobe disease seen in smokers. Patients usually present with increasing dyspnoea and weight loss, with cor pulmonale and polycythaemia occurring late in the course of the disease. Chest radiographs and high resolution CT scans typically show bilateral basal emphysema with paucity and pruning of the basal pulmonary vessels (fig1B). Upper lobe vascularisation is relatively normal and ventilation perfusion radioisotope scans and angiography also show abnormalities with a lower zone distribution.13 Lung function tests are typical for emphysema with a reduced FEV1/FVC ratio, evidence of air trapping and a low gas transfer factor.
Structure and function of α1-antitrypsin
Alpha1-antitrypsin is the archetypal member of theserine proteinase inhibitor or serpin superfamily.14 ,15 Members of the family have more than 30% sequence homology with α1-antitrypsin and share a similar molecular architecture14 based on a dominant A β-pleated sheet and nine α-helices. This structure supports an exposed, mobile, reactive loop peptide that acts as a pseudosubstrate to inhibit the target proteinase. In the case of α1-antitrypsin the loop presents a methionine residue at the P1 site to interact with the substrate binding pocket of neutrophil elastase. After binding the proteinase is inactivated by complex formation and cleared from the circulation.16Crystallographic analysis of the serpins has yielded a series of structures which illustrate the striking mobility of the reactive centre loop, allowing its function to be likened to that of a mousetrap (fig 2A). More recently our crystal structures of intact α1-antitrypsin17 ,18 have revealed the conformation of the loop that is ideal for docking with neutrophil elastase (fig 2B) before it is locked into a stable complex by the mousetrap mechanism. This mobility of the loop is essential for inhibitory function but may allow aberrant conformations by insertion into the A β-sheet of a second α1-antitrypsin molecule to form a dimer which then extends to form a chain of loop-sheet polymers.19-21 Such polymers form following treatment with mild denaturants22 and upon heating α1-antitrypsin21 and other serpins23 at high temperatures.

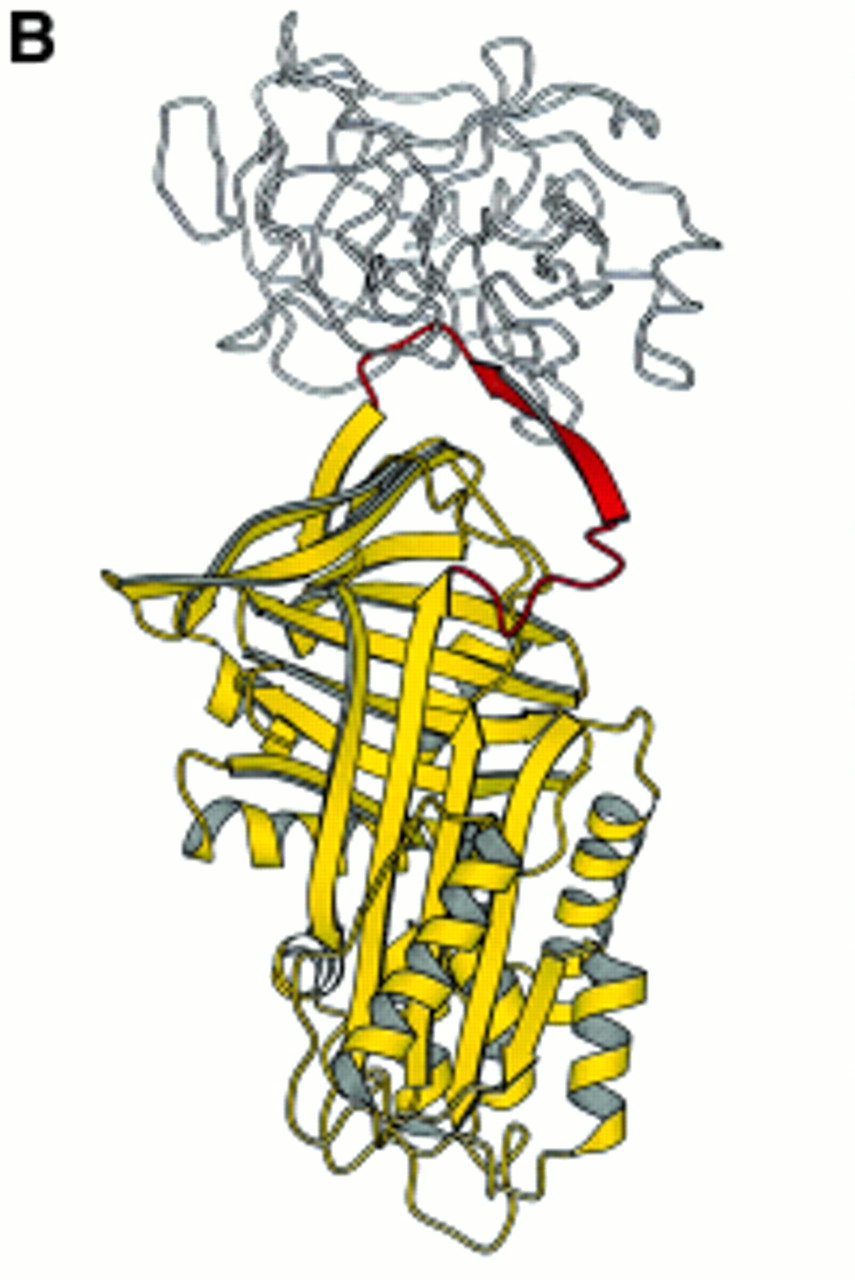
(A) The conformational transition of the serine proteinase inhibitors (serpins) modelled on antithrombin by J Whisstock and A Lesk (Department of Haematology, University of Cambridge). The mechanism by which the reactive loop moves to inhibit its target enzyme may be likened to the function of a mousetrap. The top frame shows the active circulating serpin identified in the crystal structure of antithrombin.60 ,61 Following binding to the target proteinase (not shown), the loop (shown in pink) is presumed to insert into the dominant A β-pleated sheet (shown in purple) to form the locking conformation (middle frame) that irreversibly binds and inactivates the enzyme. If antithrombin or α1-antitrypsin are heated to high temperatures in the presence of stabilising agents, the loop is able to pass around the C-sheet spur (in blue) to become fully incorporated into the molecule60 to form the latent conformation (lower frame). This conformation can be restored to the active moiety by refolding from denaturants.22 The position of the Z mutation (P17) of α1-antitrypsin is shown. (B) α1-Antitrypsin (yellow) traps the enzyme neutrophil elastase (white) to form an irreversible complex which is then cleared from the circulation. Such a complex protects the lungs against uncontrolled proteolytic degradation by neutrophil elastase (reproduced from ref 17 with permission).
Loop-sheet polymers and liver disease
The Z mutation of α1-antitrypsin is located at residue P17 (17 residues proximal to the P1reactive centre) at the head of strand 5 of the A β-sheet and the base of the mobile reactive centre loop (fig 2). We predicted that the mutation would act to open the A β-sheet thereby favouring spontaneous reactive loop into β-sheet polymerisation. Indeed, functional studies showed that Z α1-antitrypsin was more unstable24 ,25 and able to polymerise at 37°C under physiological conditions.26 The rate was accelerated by raising the temperature to 41°C and could be blocked by exogenous reactive loop peptides that compete with the loop for A β-sheet annealing. The role of this phenomenon in vivo was clarified when it was found that polymers with identical microscopic appearance could be isolated from the liver of a Z α1-antitrypsin homozygote. Although many α1-antitrypsin deficiency variants have been described, only two other mutants of α1-antitrypsin have similarly been associated with plasma deficiency and hepatic inclusions: α1-antitrypsin Siiyama (53Ser→Phe)27 and α1-antitrypsin Mmalton (52Phe deleted).28 Both of these mutants also formed spontaneous loop-sheet polymers in vivo (fig 3A).29 ,30 The temperature and concentration dependence of polymerisation, along with genetic factors,31 ,32 may account for the heterogeneity in clinical features. As α1-antitrypsin is an acute phase protein the concentration will rise during episodes of inflammation. At these times the formation of polymers is likely to overwhelm the degradative pathway, thereby exacerbating hepatic inclusions and the associated hepatocellular damage.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
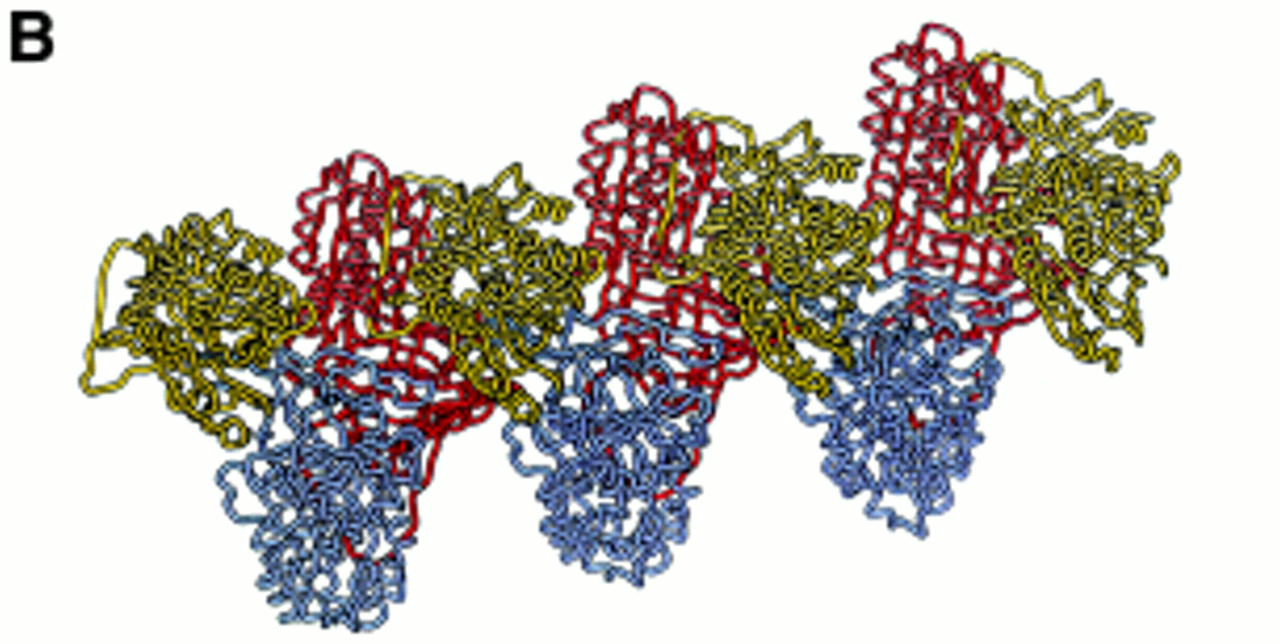
(A) Loop-sheet polymers of α1-antitrypsin isolated from the plasma of a Siiyama α1-antitrypsin homozygote. This point mutation favours the formation of long chains of polymers as well as circlets (inset). These polymers tangle in the liver to form insoluble inclusions that predispose to liver damage (reproduced from ref 29 with permission). (B) Our recent crystal structure of α1-antitrypsin provides strong support for the loop-sheet linkage being between the β-pleated reactive centre loop of one molecule and the A β-pleated sheet of the next. These dimers extend to form long chains shown here as alternating yellow, blue and red α1-antitrypsin molecules (reproduced from ref 17 with permission). The prevention of this linkage would block polymerisation and so attenuate the liver disease associated with the Z mutation.
The phenomenon of loop-sheet polymerisation is not restricted to α1-antitrypsin and has now been reported to account for the deficiency of mutants of C1 inhibitor33 and antithrombin34 ,35 in patients with angio-oedema and thrombosis, respectively, and has been linked to a deficiency of α1-antichymotrypsin in patients with liver disease and emphysema.36 ,37 It also underlies the plasma deficiency associated with common S variant of α1-antitrypsin.38 The rate of polymerisation of S α1-antitrypsin is slower than that associated with the Z variant which is compatible with the milder clinical phenotype. The precise linkage underlying the formation of polymers in vivo has been probed by site directed mutagenesis and protein expression in theXenopus oocyte system. A point mutation in the region of opening of the A sheet (51Phe→Leu) stabilises the α1-antitrypsin molecule39 and attenuates the effect of the Z mutation in vitro40 and in vivo.41 Moreover, this mutation completely prevents the accumulation of α1-antitrypsin Siiyama in aXenopus oocyte expression system.41Recombinant α1-antitrypsin with and without the51Phe→Leu mutation was expressed in E coliand purified to homogeneity. The proteins function normally as inhibitors of neutrophil elastase17 and were used for crystallographic analysis. Crystals of recombinant protein were grown and two structures of α1-antitrypsin solved to 2.9 Å. These structures,17 ,18 and that of Ryu et al,42 provide an explanation for the ready formation of loop-sheet polymers as the reactive loop strand configuration fits easily into the A β-pleated sheet of α1-antitrypsin to form a dimer that can then extend as trimers, tetramers, and so on, to as many as 20 molecules in length (fig 3B). It is these filaments which then tangle within the endoplasmic reticulum of the hepatocyte to form the insoluble inclusions that are characteristic of Z α1-antitrypsin deficiency.
Loop-sheet polymers and emphysema
Alpha1-antitrypsin deficiency related emphysema is believed to result from the reduced levels of circulating proteinase inhibitor being unable to protect the lungs against proteolytic attack.10 ,43 The Z α1-antitrypsin that does escape from the liver into the circulation is less efficient at protecting the tissues from enzyme damage21 ,44 ,45 and, like M α1-antitrypsin,46 ,47 may be inactivated by oxidation of the P1 methionine residue. The demonstration that Z α1-antitrypsin can undergo a spontaneous conformational transition in association with liver disease raised the possibility that this may also occur within the lung. Indeed, we have detected polymers in broncholaveolar lavage fluid in two out of five patients with Z α1-antitrypsin deficiency but not in lavage fluid from 13 MZ, MS, or M α1-antitrypsin controls.48 This may have important implications for the pathogenesis of disease as polymerisation obscures the reactive loop of α1-antitrypsin, rendering the protein inactive as an inhibitor of proteolytic enzymes.21 Thus, the spontaneous polymerisation of α1-antitrypsin within the lung will exacerbate the already reduced antiproteinase screen, thereby increasing the susceptibility of the tissues to proteolytic attack. The relationship of α1-antitrypsin polymers to smoking, infection, and rate of decline in lung function remains to be established and other studies are required to determine their effects on neutrophils which are found in increased numbers in the lungs of patients with α1-antitrypsin deficiency.49
Treatment
This new understanding of the structural basis of α1-antitrypsin deficiency provides a platform for rational drug design to block polymerisation in vivo and so attenuate the associated liver disease.18 Any treatment that improves secretion from the liver will raise the circulating levels of α1-antitrypsin and so enhance the antiproteinase protection within the lung. Until that time the prevention of emphysema is better than cure and there is good evidence that many Z α1-antitrypsin homozygotes would develop only mild lung disease if they abstained from smoking.50-52 The genetic deficiency in the anti-elastase screen may be rectified biochemically by intravenous infusions of α1-antitrypsin53but the role of this treatment in the prevention of emphysema is unproven. A theoretical option is the intravenous administration of genetically engineered α1-antitrypsin with the P1 methionine mutated to a valine residue.54This has little effect on the function of the protein but makes it resistant to oxidation, thereby improving its anti-elastase activity. Other treatments at an early stage of development include gene therapy and retinoic acid. Vectors carrying the α1-antitrypsin gene have been targeted to liver55 ,56 and lung57 but there is currently insufficient gene expression for this to be a useful therapeutic modality. Similarly, although the effects of retinoic acid on alveolar regeneration in the rat look promising,58 it has yet to be tested in man.
In the meantime patients with α1-antitrypsin deficiency related emphysema should receive conventional therapy with trials of bronchodilators, inhaled corticosteroids and, where appropriate, assessment for long term oxygen therapy and single lung transplantation. The role of lung volume reduction surgery in this group is even more contentious than in smoking related centrilobular emphysema as the disease is basal rather than apical and typically lacks the target areas which are most suitable for resection.
Conclusion
Z α1-antitrypsin deficiency results from the accumulation of loop-sheet polymers within the endoplasmic reticulum of the hepatocyte. These polymers also form within the lung where they inactivate α1-antitrypsin, thereby further reducing the antiproteinase screen that protects the tissues against proteolytic attack. Recent advances in understanding the transition from monomeric to polymeric protein raises the prospect of rational drug design to treat the deficiency and provides a useful paradigm for other conformational diseases such as the prion diseases, amyloidosis and Alzheimer’s disease.59