Article Text

Abstract
Background: This study was conducted to determine whether the major nasal airway ion transport abnormalities in cystic fibrosis (that is, defective cAMP regulated chloride secretion and basal sodium hyperabsorption) are related to the clinical expression of cystic fibrosis and/or to the genotype.
Methods: Nasal potential difference was measured in 79 adult patients with cystic fibrosis for whom clinical status, respiratory function, and CFTR genotype were determined.
Results: In univariate and multivariate analysis, patients with pancreatic insufficiency were more likely to have low responses to low chloride (odds ratio (OR) 8.6 (95% CI 1.3 to 58.5), p = 0.03) and isoproterenol (OR 11.2 (95% CI 1.3 to 93.9), p = 0.03) solutions. Similarly, in univariate and multivariate analysis, patients with poor respiratory function (forced expiratory volume in 1 second <50% of predicted value) were more likely to have an enhanced response to amiloride solution (OR 3.7 (95% CI 1.3 to 11.0), p = 0.02). However, there was no significant relationship between nasal potential difference and the severity of the genotype.
Conclusions: Nasal epithelial ion transport in cystic fibrosis is linked to the clinical expression of the disease. The pancreatic status appears to be mostly related to the defect in epithelial chloride secretion whereas the respiratory status is mostly related to abnormal sodium transport and the regulatory function of the CFTR protein.
- nasal potential difference
- cystic fibrosis
- ion transport
- genotype/phenotype
Statistics from Altmetric.com
The cystic fibrosis transmembrane conductance regulator (CFTR) protein plays a key role in the electrolyte transport across the epithelium via its function as a cAMP dependent chloride channel1,2 and its regulatory roles on other chloride and sodium channels.3–5 The two major ion transport abnormalities in cystic fibrosis (CF) respiratory epithelium are a defect in cAMP regulated chloride secretion and an acceleration of the rate of basal sodium absorption.6–8 Each component of the airway epithelial ion transport can be assessed easily in vivo by measuring the nasal potential difference (PD) under different conditions.9–12 Increased sodium absorption is reflected in an increase in basal nasal PD together with an increased response to the sodium channel blocker amiloride. The defect in chloride movement is demonstrated after perfusion with a low chloride solution in the presence of amiloride and subsequent perfusion with the cAMP increasing agent isoproterenol. Both agents induce a small and transient response in CF patients which is in contrast to the rapid and sustained response observed in non-CF subjects.
As nasal PD reflects the expression and function of the various airway ion channels including CFTR, it might be associated with CF clinical outcome. We have previously shown in a group of 95 adult patients with CF that patients with the lowest basal nasal PD had better respiratory function, irrespective of the severity of the genotype.13 Other groups have suggested some correlation between the severity of lung disease and cAMP mediated chloride secretion.14,15 Moreover, in 114 twins and siblings homozygous for the F508Δ mutation, the most common CFTR mutation, the expression of basal chloride conductance was identified as a positive predictor of milder respiratory disease.16 In contrast, in 51 young CF patients, a recent study showed no correlation between any component of ion transport measurement and respiratory function.17 In the present study we examined whether some components of nasal epithelial ion transport were associated with the pancreatic and respiratory expression of the disease and/or the genotype in adults with CF.
METHODS
Patients
Seventy nine adult patients with CF who regularly attended the adult CF centre at Cochin Hospital, Paris, France between 1 January 2000 and 30 June 2003 were included in the study. A definitive diagnosis of CF was confirmed by two abnormal sweat chloride measurements (⩾60 mmol/l) and/or the identification of two mutations in the CFTR gene. Exclusion criteria included acute rhinitis over the previous 4 weeks, nasal polyposis, topical or oral steroids, and long term nasal oxygen therapy. Clinical assessment and nasal PD studies were undertaken when patients were in a stable condition. Pancreatic status was determined by measuring the fat or elastase content of stool samples collected over 3 days: patients with faecal fat <6 g/day and currently not treated by pancreatic enzyme replacement or with faecal elastase above 200 μg/g faeces were defined as pancreatic sufficient, and the others as pancreatic insufficient. Values of forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) were determined on the day of nasal PD measurements and expressed as percentages of predicted values. Arterial oxygen and carbon dioxide tensions (Pao2, Paco2) were measured at rest.
The study was approved by the ethical committee of our hospital.
Nasal PD measurement
Nasal PD was measured by a technician blind to the clinical status of the patients. The method, based on two silver/silver chloride electrodes connected to a high impedance voltmeter, was adapted from Alton et al.10 The following nasal PD measurements were recorded: (1) the stable maximal baseline PD; (2) the response to superfusion with 10−4 M amiloride, a blocker of epithelial sodium channel (Δamil); (3) the change in PD following a perfusion of a low chloride solution (ΔCl−/amil) to allow the determination of basal chloride conductance (in this solution the chloride contained in the amiloride solution was replaced by gluconate); and (4) the response to the β adrenergic agonist isoproterenol (10−5 M) (Δiso/Cl−) which was added to the low chloride solution containing amiloride. Isoproterenol induces cAMP dependent chloride conductance and determines the presence of CFTR. Superfusion of the nasal epithelium with each solution was continued until a steady state was reached, or for at least 3 minutes.
Twenty six non-CF subjects with bronchiectasis were studied to determine control values. All subjects had a normal sweat test and no mutation in the CFTR gene. The median (range) results were as follows: basal nasal PD –18 mV (–30 to –11); Δamil 13 mV (2–26); ΔCl−/amil –12 mV (–36 to –5); Δiso/Cl− –6 mV (–15 to 7) (fig 1).

Nasal PD measurements in CF patients and controls. Basal PD was measured with a Kreb’s HEPES solution, after which the responses to superfusion with 10−4 M amiloride (Δamil), a low chloride solution (ΔCl−/amil) and isoproterenol (10−5 M) (Δiso/Cl−) were recorded. Each box plot is composed of five horizontal lines that display the 10th, 25th, 50th, 75th and 90th percentiles of the variable. All values for the variable above the 90th percentile and below the 10th percentile are plotted separately.
Deoxyribonucleic acid (DNA) analysis and classification of mutations
For all patients, all 27 exons and flanking regions of the CFTR gene were screened for mutations, either with a commercially available kit (Cystic Fibrosis Assay, Applied Biosystems, Foster City, CA, USA) for the 31 most frequent mutations in the CFTR gene or, for totally or partially uncharacterised samples, by denaturing gradient gel electrophoresis (DGGE) on genomic DNA amplified by the polymerase chain reaction.18 Patients were classified into two genotype groups according to the probable effect of their mutations on CFTR function, regardless of clinical features, using the classification of CFTR mutations proposed by Welsh and Smith19 and expanded by Zielenski and Tsui.20 The “severe” genotype group included patients with two CFTR mutations belonging to class I, II or III, while the “mild” genotype group included patients with at least one mutation belonging to class IV or V.
Statistical analysis
Continuous variables were expressed as median (range). Univariate correlations between continuous variables were explored using Spearman’s rank correlation. Comparisons were tested using Spearman’s rank correlation test or the non-parametric Mann-Whitney U test. Multivariate analysis of factors associated with pancreatic status, respiratory function, and genotype were performed as follows. All continuous variables were dichotomised according to the median or to a cut off point of 50% of the predicted value for FEV1. The cut off point of 50% of the predicted value for FEV1 was chosen because it is usually the cut off point below which an obstructive abnormality is considered “severe”.21 A univariate analysis was first performed using the Fisher’s exact test. Thus, multivariate logistic regression models were built first including variables when the univariate p value was less than 0.2 and finally keeping variables in the model by a backward stepwise regression and by maximising the likelihood of the model with the likelihood ratio test. Stata/SE 8.0 software was used. p values of ⩽0.05 were considered statistically significant.
RESULTS
Patients
Four of the 79 CF patients included in the study had a normal sweat test; three were compound heterozygous for the F508Δ mutation and the R117H, D1152H and R347H mutations, respectively, and one patient was compound heterozygous for the G542X and 3849+10 kb (C)→ (T) mutations. The sweat chloride levels for these patients were 40, 13, 49 and 52 mmol/l, respectively. All patients had bronchiectasis confirmed by computed tomographic (CT) scanning. Table 1 shows the clinical characteristics of the patients. In six patients only one mutation was identified. The 73 patients for whom two mutations were identified were classified into two genotype groups: the “severe” genotype group included 51 patients of whom 25 were homozygous for the F508Δ mutation, and the “mild” genotype group comprised 22 patients.
Clinical and genotype data of the CF patients
Nasal PD measurements
Nasal PD measurements in the 79 CF patients are shown in table 1 and fig 1. They were different from those observed in our non-CF control group: CF patients had increased sodium transport, as shown by increased basal PD and increased response to amiloride perfusion (p<0.0001 for both), and a low response to perfusion with low chloride solution and isoproterenol (p<0.0001 for both; fig 1). Six CF patients had, in absolute value, a basal PD of ⩽31 mV which represents the mean+2SD of our control group. However, all had a typical CF response to pharmacological solutions with an absence or a very low response to low chloride solution, isoproterenol, or both.
Relationships between nasal PD, phenotype and CFTR genotype
No difference in sodium transport was observed between patients with pancreatic sufficiency or insufficiency. However, patients with pancreatic sufficiency had a higher response to low chloride solution and isoproterenol than patients with pancreatic insufficiency (p = 0.003 and p = 0.01, respectively, table 2, figs 2 and 3).
Nasal PD measurements, clinical data and genotype: univariate analysis

Pancreatic status and epithelial chloride secretion in CF patients. Responses to superfusion with a low chloride solution (ΔCl−/amil) and isoproterenol (10−5 M) (Δiso/Cl−) are shown in patients with pancreatic sufficiency (PS) and insufficiency (PI). Each box plot is composed of five horizontal lines that display the 10th, 25th, 50th, 75th and 90th percentiles of the variable. All values for the variable above the 90th percentile and below the 10th percentile are plotted separately.
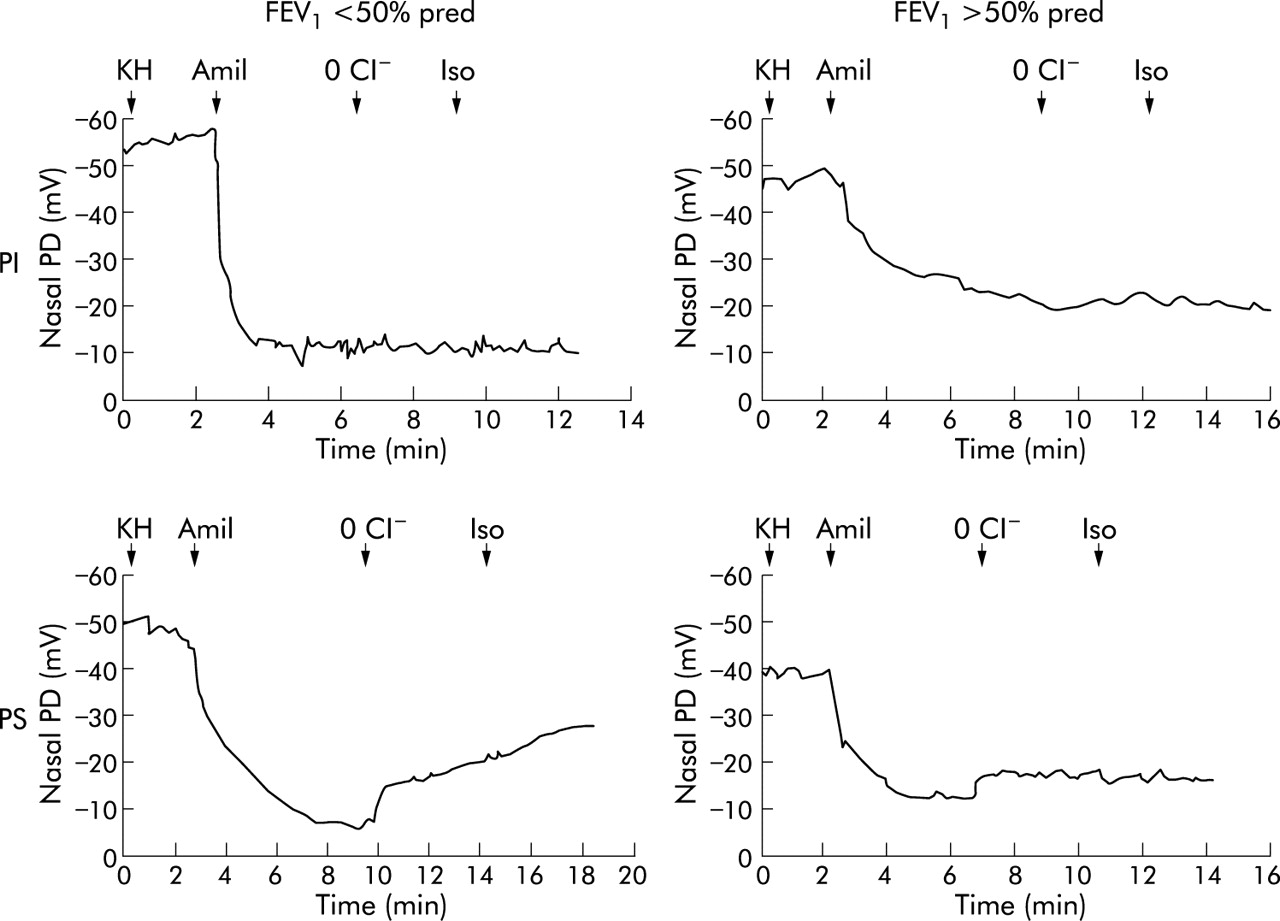
Nasal PD tracings in four patients with CF. Basal PD was measured with a Kreb’s HEPES solution (KH), after which the nasal epithelium was superfused with 10−4 M amiloride (Amil), a low chloride solution (0 Cl−), and 10−5 M isoproterenol (Iso). Tracings are shown for two CF patients with pancreatic insufficiency (top panels) and FEV1 <50% pred (left panel) or FEV1 >50% pred (right panel), both of whom were homozygous for the F508Δ mutation and belonged to the “severe” genotype group; and two CF patients with pancreatic sufficiency (bottom panels) and FEV1 <50% pred (left panel) or FEV1 >50% pred (right panel), both of whom were compound heterozygous for the F508Δ mutation and the R117H and G85E mutations, respectively, and belonged to the “mild” genotype group. The two patients with FEV1 <50% pred had a higher basal nasal PD and a higher response to amiloride than the two patients with FEV1 >50% pred, and the two patients with pancreatic sufficiency had a higher response to low chloride solution and isoproterenol than the patients with pancreatic insufficiency.
There was no difference in basal PD and in the response to the different solutions between patients with or without P aeruginosa colonisation (table 2). Basal nasal PD and responses to amiloride were both correlated with FEV1 (p = 0.04 and p = 0.009, respectively, table 2) and FVC (p = 0.03 and p = 0.01, respectively, table 2). When FEV1 was classified into two groups of severity with a cut off point of 50% of the predicted value, severe respiratory function was similarly associated with a high basal nasal PD and a high response to amiloride (p = 0.005 and 0.003, respectively, table 2, figs 3 and 4). No correlation was observed between the indicators of pulmonary function and the responses to low chloride solution or isoproterenol, except for Paco2 and the response to isoproterenol (p = 0.04, table 2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Respiratory status as assessed by FEV1 and epithelial sodium absorption in patients with CF. The basal PD and response to superfusion with 10−4 M amiloride (Δamil) are shown in patients with FEV1 ⩾50% and <50% of the predicted value (pred). Each box plot is composed of five horizontal lines that display the 10th, 25th, 50th, 75th and 90th percentiles of the variable. All values for the variable above the 90th percentile and below the 10th percentile are plotted separately.
No relationship was observed between nasal PD and all other clinical characteristics (sex, age and symptoms at diagnosis, sweat chloride concentrations, BMI, the presence of meconium ileus or diabetes). Patients with the “mild” genotype had a lower basal nasal PD than those with the “severe” genotype (p = 0.01, table 2). No difference was observed between the severity of the genotype and the responses to amiloride, low chloride solution, or isoproterenol.
Factors associated with a severe phenotype or a severe genotype
In order to evaluate which data were associated with the pancreatic status, quantitative data were classified into two groups of severity and a multivariate analysis was performed (table 3). Patients with pancreatic insufficiency were more likely to have a “severe” genotype (odds ratio (OR) 28.2 (95% confidence interval (CI) 4.5 to 178.1), p<0.001), a “low” BMI (OR 8.8 (95% CI 1.3 to 58.3), p = 0.03), and low responses to low chloride (OR 8.6 (95% CI 1.3 to 58.5), p = 0.03) and isoproterenol (OR 11.2 (95% CI 1.3 to 93.9), p = 0.03) solutions.
Analysis to determine which parameters are associated with pancreatic insufficiency
In order to evaluate which data were associated with the respiratory function as assessed by FEV1, a similar analysis was performed (table 4). Patients with FEV1 <50% predicted were more likely to be colonised with P aeruginosa (OR 13.8 (95% CI 2.8 to 68.6), p = 0.001) and to have an enhanced response to amiloride solution (OR 3.7 (95% CI 1.3 to 11.0), p = 0.02).
Analysis to determine which parameters are associated with respiratory insufficiency as assessed by FEV1 <50% of predicted value
Using a similar multivariate analysis we studied which data were associated with the genotype. There was no significant relationship between nasal PD measurements and the severity of the genotype. The only parameter linked to the “severe” genotype group was pancreatic insufficiency (OR 15.7 (95% CI 4.0 to 61.6), p<0.0001).
DISCUSSION
In this study we have shown that the basic defects in CF—that is, the two major ion transport abnormalities—are associated with the two major clinical components of the disease: pancreatic insufficiency and lung disease. The pancreatic status is mostly related to epithelial chloride secretion whereas the severity of lung disease is linked to abnormal sodium transport.
In univariate analysis the pancreatic status was related to both basal and cAMP regulated chloride secretion. This was confirmed by the multivariate analysis which showed that patients with pancreatic insufficiency were more likely to have a severe genotype, low BMI, and decreased basal and cAMP regulated chloride permeabilities across the nasal airway epithelium. The relationship between the genotype and the pancreatic status is well established.22 Similarly, the link between exocrine pancreatic insufficiency causing maldigestion and malabsorption of fats and proteins and a low BMI is obvious. In this study we show that there is also a link between nasal airway chloride secretion and pancreatic status. A relationship between residual chloride transport in intestinal tissue and a mildly affected phenotype, as assessed by the predicted weight for height and the FEV1, has previously been described in F508Δ homozygous twins and siblings by Bronsveld et al16 but the reverse—that is, the relationship between nasal airway ion transport and the nutritional status—was not examined by these researchers nor in a recent study of 51 young CF patients of whom only three were pancreatic sufficient.17
The relationship we have observed after both low chloride and isoproterenol solutions may reflect the fact that the pancreatic status in CF is related to the residual function of CFTR. Such a relationship is in keeping with the well known relationship between CFTR genotype and pancreatic status. CF pancreatic insufficiency is thought to be due mainly to abnormal bicarbonate secretion in which CFTR plays a critical but poorly defined role.23 In epithelial cells the cAMP stimulated chloride current is conducted by both CFTR and the outwardly rectifying chloride channel.24 Hence, the relationship we observed between responses to low chloride and isoproterenol solutions and pancreatic status raises the question of a possible role for other chloride channels regulated by CFTR in CF pancreatic insufficiency.
Univariate analysis showed that, in adults with CF, irrespective of the severity of the genotype, the severity of the respiratory function is related to the extent of sodium hyperabsorption as assessed by basal nasal PD and response to the sodium blocker amiloride. In order to perform a multivariate analysis, the respiratory function, as assessed by FEV1, was classified into two groups of severity according to a cut off point of 50% of the predicted value. FEV1 was chosen because it is the variable that best reflects the status of lung function throughout the course of CF lung disease,25,26 and the cut off point of 50% of the predicted value is usually the point below which an obstructive abnormality is considered “severe”.21 Multivariate analysis showed that the severity of the respiratory function as assessed by FEV1 is linked to P aeruginosa colonisation, a well known factor linked to poor respiratory prognosis, but also to the magnitude of the response to amiloride. In multivariate analysis, respiratory function was not linked to basal nasal PD, the other index of sodium transport rate. This discrepancy, which was not found in univariate analysis, is likely to be due to the small sample size. The relationship we found between respiratory function and sodium transport confirms and expands our previously published data showing that, in adult CF patients, normal sodium absorption is related to milder respiratory disease, irrespective of the severity of the genotype.13 In a recent study Wallace et al did not observe any relationship between nasal airway ion transport and lung function.17 However, this study was conducted in 51 young CF patients which included only three patients with an FEV1 less than 50% of the predicted value, making it difficult to draw conclusions on the influence of ion transport on the severity of lung disease.
In contrast to our results, Bronsveld et al found that, in 114 F508Δ homozygous twins and siblings, the expression of basal chloride conductance was a positive predictor of milder respiratory disease.16 The discrepancies between our study and that of Bronsveld et al might be due to the different study populations: the respiratory function appears to be related to chloride transport in a highly selected and defined population such as the one studied by Bronsveld et al.16 However, in a population with a wide range of genotypes such as ours, the pulmonary outcome appears to be mostly influenced by increased sodium absorption in the airway epithelium. These results suggest that the severity of pulmonary disease is more closely associated with the regulatory than with the chloride channel function of CFTR. They are in accordance with the “low airway surface liquid volume hypothesis” currently proposed as one possible mechanism for CF airway disease which states that hyperabsorption of sodium observed in CF would result in a viscous and dehydrated airway surface liquid that impairs mucociliary function and facilitates bacterial adherence and colonisation.27 They are also in accordance with recent data showing that transgenic mice overexpressing the β subunit of the epithelial sodium channel exhibit a CF-like lung disease.28
In univariate analysis the severity of the genotype was linked to defective nasal sodium transport. This was not confirmed by multivariate analysis in which, as previously reported,22 the genotype was only linked to the pancreatic status. A number of studies have found an association between some genetic variants and respiratory function in CF. In particular, polymorphisms affecting the function of genes that mediate innate immunity and other inflammatory processes have been highlighted.29 Our data further emphasise that factors other than the CFTR gene contribute to airway epithelial ion transport measured by nasal PD. They also point to the importance of measuring nasal PD in patients with CF since it reflects not only CFTR function and its regulation of other channels but also the impact of other genetic or environmental factors affecting the disease phenotype.
We conclude that, in a population of CF adults, the clinical outcome is associated with expression of the basic ion transport defects. Pancreatic insufficiency is linked to highly defective epithelial chloride secretion, whereas the severity of lung disease is associated with the extent of sodium hyperabsorption. This is one of the few reports to link the major ion transport abnormalities in CF with the major clinical components of the disease.13–16 These findings suggest that measurement of nasal PD is useful not only for CF diagnosis, but also to gain more insight into the patient’s electrophysiological characteristics in correlation with disease severity.
Acknowledgments
The authors thank S Remeur, M Lachaux, S Rebeyrol, M Morin and C Bessière (Service d’Explorations Fonctionnelles, Hôpital Cochin, Paris, France) for technical assistance and C Bernède (CeRBP, Institut Pasteur, Paris, France) for statistical assistance.
REFERENCES
Footnotes
-
This study was supported by the Association Vaincre la Mucoviscidose.