Article Text

Abstract
Background: The alveolar compartment is a procoagulant antifibrinolytic environment in acute lung injury (ALI) and the acute respiratory distress syndrome (ARDS). A study was undertaken to test the hypothesis that the alveolar epithelium can initiate intra-alveolar coagulation by expressing active tissue factor (TF).
Methods: Using an in vitro cell surface TF assay and TF ELISA, the activity and production of TF in cultured alveolar epithelial (A549) cells following exposure to cytomix (tumour necrosis factor α, interleukin 1β and interferon γ) was measured. TF gene transcription was measured by semi-quantitative reverse-transcription PCR. Immunohistochemistry for TF was performed on lung sections from patients with ARDS and controls. TF protein levels were measured by ELISA in undiluted pulmonary oedema fluid from patients with ALI/ARDS and compared with control patients with hydrostatic pulmonary oedema.
Results: TF activity, mRNA and protein levels increased in A549 cells after stimulation with cytomix. Increased TF activity was also seen in A549 cells following incubation with pulmonary oedema fluid from patients with ALI/ARDS. Immunohistochemistry for TF in human lung tissue from patients with ARDS showed prominent TF staining in alveolar epithelial cells as well as intra-alveolar macrophages and hyaline membranes. TF antigen levels in oedema fluid (median 37 113 (IQR 14 956–73 525) pg/ml) were significantly higher than in plasma (median 336 (IQR 165–669) pg/ml, p<0.001) in patients with ALI/ARDS, and TF procoagulant activity in oedema fluid was much higher than in plasma of these patients. Higher plasma levels were associated with mortality.
Conclusions: The alveolar epithelium is capable of modulating intra-alveolar coagulation through upregulation of TF following exposure to inflammatory stimuli and may contribute to intra-alveolar fibrin deposition in ARDS.
- ALI, acute lung injury
- ARDS, acute respiratory distress syndrome
- BAL, bronchoalveolar lavage
- Ct, confidence threshold
- PMA, phorbol myristate acetate
- MEM, minimal essential medium
- SAEC, small airway epithelial cells
- TF, tissue factor
Statistics from Altmetric.com
- ALI, acute lung injury
- ARDS, acute respiratory distress syndrome
- BAL, bronchoalveolar lavage
- Ct, confidence threshold
- PMA, phorbol myristate acetate
- MEM, minimal essential medium
- SAEC, small airway epithelial cells
- TF, tissue factor
Intra-alveolar fibrin deposition is a pathological hallmark of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS).1 Although the alveolar compartment in ALI/ARDS is procoagulant and antifibrinolytic,2,3 the cellular and molecular mechanisms that modulate intra-alveolar fibrin deposition are incompletely understood. Previous studies have shown that there is TF-dependent procoagulant activity in bronchoalveolar lavage (BAL) fluid in patients with ARDS,2,4 but comparison with a comparable control group (intubated patients with hydrostatic oedema) was not performed. The potential importance of the coagulation and fibrinolytic pathways in the alveolar space in ALI/ARDS needs further assessment. Clinical trials in ALI/ARDS have primarily targeted inflammation and have not focused on coagulation abnormalities. The recent success of an anticoagulant therapy in reducing mortality in severe sepsis,5 as well as progress in basic research,6 highlights the important pathogenetic role of coagulation abnormalities in inflammatory disorders.
Tissue factor (TF) is a 47 kDa transmembrane glycoprotein that is the most potent stimulator of the extrinsic coagulation cascade. Factor VIIa binds to TF on the cell surface and this complex binds to factor X, converting it to the activated form Xa which leads to eventual thrombin formation and fibrin deposition. Idell et al7 reported that TF activity correlated temporally with alveolar fibrin deposition in BAL fluid in a baboon model of ALI, and that TF was an important contributor to the procoagulant activity in BAL fluid from patients with ALI/ARDS.2 Carraway et al8 demonstrated the central role of TF in sepsis-induced organ dysfunction (including lung injury) in an E coli model of septic shock. In that study, baboons treated with site inactivated VIIa, which blocks TF activity, had a dramatic reduction in physiological and histological lung injury as well as improved survival.8 Similar findings were reported using a blocking antibody for the TF-factor X interaction in the same model.9 Several studies have demonstrated the direct link between inflammation and TF activation.10–12 Because of its central role in both fibrin generation and in linking inflammation and coagulation, TF may have a central role in the pathogenesis of ALI/ARDS.
Although both alveolar macrophages and alveolar epithelial cells have been identified as potential sources of alveolar TF, the specific time course and mechanism of alveolar epithelial TF production has not been well studied. Freshly isolated alveolar epithelial cells from rats can express TF, but only one study showed a decrease in TF activity with phorbol myristate acetate (PMA) stimulation13 while another demonstrated an increase in TF procoagulant activity.14 In addition, detailed information about the mechanism of TF expression by human alveolar epithelial cells is lacking. We hypothesised that active TF is present in the alveolar compartment of patients with ALI/ARDS compared with patients with hydrostatic pulmonary oedema and that the alveolar epithelium is a cellular source for soluble TF. We also hypothesised that the alveolar epithelium can express active TF and thus can actively modulate intra-alveolar coagulation and fibrin deposition.
METHODS
Collection of pulmonary oedema fluid
Samples of human pulmonary oedema fluid and plasma were collected as previously described from patients in the intensive care units at Moffitt Long and San Francisco general hospitals from 1995 to 2001.15 Briefly, mechanically ventilated patients with ALI/ARDS or controls with hydrostatic pulmonary oedema were identified within 6 h of endotracheal intubation using standard definitions.16 A standard tracheal suction catheter was inserted through the endotracheal tube and advanced slowly until resistance was met. Gentle suction was applied and a few millilitres of undiluted pulmonary oedema fluid were aspirated. Samples were placed immediately on ice and centrifuged for 10 min at 3000 g to remove any cellular or particulate matter. Supernatants were stored at –70°C until use. A simultaneous plasma sample was collected from each patient.
The study was approved by the Committee on Human Research at the University of California San Francisco (UCSF). The Institutional Review Board at UCSF allowed a waiver of informed consent as the samples obtained were part of routine care and would otherwise have been discarded.
Tissue factor enzyme-linked immunosorbent assay (ELISA)
A commercially available ELISA kit for TF (American Diagnostica, Stamford, Connecticut, USA) was used to measure protein concentrations of TF in human samples, cell lysates and supernatants according to the manufacturer’s instructions. All measurements were done in duplicate.
Effect of pulmonary oedema fluid on clot formation in normal plasma
Clot time was measured using a mechanical clot detection system (Start 4 Coagulometer, Diagnostica Stago, Asnieres, France). Briefly, pulmonary oedema fluid was diluted 1:20 in phosphate buffered saline (PBS) to a volume of 50 μl and incubated for 30 min at 37°C with either a blocking antibody to TF (American Diagnostica) or a species-matched anti-human IgG antibody (Sigma, St Louis, Missouri, USA) at a dilution of 1:6. Samples were then incubated with 50 μl pooled normal plasma (Fisher Diagnostics, Middletown, Virginia, USA) for 2 min. Clot time was determined as recalcification time following the addition of 8.3 mM calcium chloride. Measurements were done in duplicate.
Immunohistochemistry study subjects
Fresh lung tissue was obtained from 12 patients admitted to the adult medical intensive care units of the University of Utah Hospital, Salt Lake City between 1999 and 2002 who died and underwent a necropsy. The necropsy tissue was collected within 12–14 h of death using standard anatomical pathology approaches optimised to preserve tissue architecture and antigen display. Necropsy tissue was obtained from two groups, each of six patients: (1) patients identified by the American European Consensus Conference definitions16 who had died with ALI or ARDS; and (2) patients who had died without clinical or pathological evidence of pulmonary disease (controls). The necropsy protocol was approved by the Institutional Review Board at the University of Utah Health Sciences Center. The requirement for written informed consent was waived.
Tissue collection and immunohistochemistry
Two to three tissue cubes (>2×2×2 cm) were obtained from each of the three lobes of the right lung of each patient. The cubes were processed by standard methods.17 Briefly, the tissue cubes were immersion-fixed in 10% buffered neutral formalin (VWR Inc, Media, Pennsylvania, USA) overnight at 4°C. Slices of the cubes were cut and processed for paraffin embedding for immunohistochemistry (6 μm section thickness).
Antigen retrieval methods were used for immunohistochemistry, followed by standard immunolabelling methods.17 Antigen retrieval was done using 1× Citra solution (BioGenex, San Ramon, California, USA). Endogenous per-oxidase was quenched by 3% hydrogen peroxide in PBS for 20 min at room temperature. Between each of the steps the tissue sections were rinsed in PBS. TF monoclonal antibody (American Diagnostica) was used at a dilution of 1:100 (overnight at 4°C). A standard ABC kit (Vector Laboratories Inc, Burlingame, California, USA) and DAB (Sigma-Aldrich Co, St Louis, Missouri, USA) were used to reveal localisation of TF. All of the tissue sections were counterstained with haematoxy-lin, dehydrated through an ethanol series, cleared in AmeriClear and covered by mounting medium and a coverslip. Digital images were obtained using a Zeiss Axiophot (Jena, Germany) equipped with a digital camera. Brightness and contrast were adjusted to match background among the digital images; however, colour adjustment and other image adjustments were not permitted.
For double immunofluorescence microscopy, deparaffinised lung sections were permeated with 1% Triton X-100, endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 20 min at room temperature, and antigen retrieval was performed using 1× Glyca antigen retrieval solution (BioGenex). Sections were blocked with 1% casein solution for 30 min at room temperature. The slides were incubated with a goat polyclonal anti-TF IgG antibody (1:500 and 1:1000 dilutions; Santa Cruz Biotechnology, Santa Cruz, California, USA) at 4°C overnight. The slides were then incubated with 1:250 anti-goat biotin conjugate (Vector Laboratories Inc) for 60 min at room temperature, followed by 1:250 streptavadin-HRP (PerkinElmer, Boston, Massachusetts, USA) for 60 min at room temperature. To visualize TF, the slides were incubated with 1:100 tyramide-FITC (PerkinElmer) for 20 min at room temperature. Sections were then incubated with a rabbit polyclonal anti-pro-Surfactant Protein B antibody (Chemicon International, Temecula, California, USA) at a dilution of 1:250 for 60 min, then with 1:100 anti-rabbit-HRP conjugate (Vector Laboratories Inc) for 60 min at room temperature, followed by tyramine-rhodamine (PerkinElmer) for 20 min at room temperature. Sections were mounted with Vectorshield mounting medium containing DAPI nuclear stain (Vector Laboratories Inc). For all experiments, three negative controls were used: isotype-matched and species-matched irrelevant antibody (anti-insulin), omission of the primary antibody and omission of the secondary antibody. In all cases the negative controls showed that the TF immunostaining was specific.
Cell culture
Alveolar epithelial cells (A549 cells, American Type Culture Collection, Manassas, Virginia, USA) were grown in MEM (Cellgro, Herndon, Virginia, USA) with 10% fetal bovine serum (Cellgro) and 10 000 units/ml each of penicillin and streptomycin (Sigma). Small airway epithelial cells (SAEC) (Clonetics, San Diego, California, USA) were grown in complete small airway growth media (Clonetics). Both cell types were subcultured at the same density. For TF activity assays, cells were plated at 1×104 cells/well in a 96-well plate and maintained in culture until confluent at 5 days. For cell lysates and RNA extraction, cells were plated in a 24-well plate at 5×104 cells/well and maintained in culture until confluence was reached at day 5. All experiments were performed on day 5 at confluence.
TF activity assay
Confluent monolayers of A549 cells were gently washed twice with 200 μl serum-free MEM then incubated for varying lengths of time with serum-free MEM with or without the addition of various concentrations of cytomix. SAEC were washed twice with small airway basal medium and TF activity was measured in the same way. Cytomix is a mixture of equal amounts of tumour necrosis factor α, interleukin 1β and interferon γ (R+D Systems, Minneapolis, Minnesota, USA). Following incubation, the cells were washed twice with 200 μl serum-free minimal essential medium (MEM), then incubated at 37°C for 60 min with 25 μl of 4 nM factor VIIa and 25 μl of 1 μM factor X. A factor Xa (factors VIIa, X, Xa, Enzyme Research Laboratories, South Bend, Indiana, USA) standard curve was prepared in a new 96-well plate. 100 μl from each sample well was transferred to the new plate. After transfer, 5 μl 100 mM EDTA was added to each well to stop the enzymatic reaction. Factor Xa was quantified by adding 100 μl of the chromogenic substrate S-2222 (Chromogenix, Milan, Italy) to each well and incubating at room temperature for 15 min followed by measurement of OD 405. Control experiments were performed without the addition of factor VIIa and/or factor X. No TF activity was measurable in the absence of added factors VIIa or X (data not shown).
Preparation of cell lysates
A549 cells were grown to confluence in 24 well plates and exposed to cytomix or control medium. SAEC were grown in small airway growth medium. Following exposure, the cells were incubated with 250 μl lysis buffer (50 mM Tris, 100 mM NaCl, 0.1% Triton X-100, pH 7.45) for 15 min at 37°C. The lysate was removed and placed in a clean microfuge tube and centrifuged at 3000 g to remove cellular debris. The supernatants were stored at –70°C until assays were performed.
Cell viability assay
Following the TF activity assay, cell viability was measured using a chromogenic electron coupling reagent (Promega, Madison, Wisconsin, USA). Cell viability was compared between wells using control wells as a reference. Cytomix stimulation did not significantly alter cell viability (data not shown). TF activity assay results are expressed as “TF index” which controls for cell viability.
Isolation of mRNA
A549 cells were cultured in 24 well plates until confluent. An appropriate stimulus was applied in serum-free MEM to cells before mRNA isolation. In addition to cytomix, some cells were also treated with actinomycin D 5 μg/ml (Sigma-Aldrich). For these experiments there were three treatment groups: untreated cells, cytomix alone and actinomycin D + cytomix (actinomycin was added 10 min before cytomix). The cells were then trypsinised and collected into cryotubes. Cells from three identical wells were pooled for each mRNA extraction. mRNA was extracted using a kit from Qiagen (Valencia, California, ISA) according to the manufacturer’s instructions and stored immediately at –70°C. SAEC were washed with small airway basal medium and RNA was isolated in the same manner.
Real-time PCR
Semi-quantitative PCR was done using mRNA isolated from A549 cells or SAEC using the SYBRgreen method (Bio-Rad, Hercules, California, USA). Before the PCR reaction, cDNA was prepared using Superscript II RT (Invitrogen, Carlsbad, California, USA). All quantitative PCR reactions were run with a no template control for each primer set and a no cDNA control from each experimental condition. Both TF and β-actin primers were obtained from IDT (Coralville, Iowa, USA) according to the specifications of Malarstig et al.18 Semi-quantitation was established using the ΔΔCt method.19 Briefly, the Ct (confidence threshold) for each well was calculated by the Bio-Rad iCycler. For each experimental condition, the Ct for β-actin was subtracted from the Ct for TF to generate the ΔCt. The difference between the ΔCt of the control experiment and the ΔCt of treated cells was calculated to give the ΔΔCt. The fold increase in mRNA was calculated by 2ΔΔCt as a quantitative estimate.
TF activity after exposure to human pulmonary oedema fluid
A549 alveolar epithelial-like cells were grown to confluence in 96-well plates. Confluent monolayers were washed with medium (RPMI) and incubated with pulmonary oedema fluid or plasma from patients with ALI/ARDS at a 1:10 dilution with RPMI for 4 h at 37°C and 5% CO2. After repeated washing to remove any Xa present in the oedema fluid, TF-dependent factor Xa activity on A549 epithelial cells was measured at 405 nm after incubation with purified factor VIIa and factor X at 2 h using a specific chromogenic substrate (S-2765).
Statistical analysis
Statistical analysis was done using SPSS V.11 for Macintosh. In the in vitro experiments, differences between groups were assessed using a one-way analysis of variance (ANOVA) followed by Tukey test for post hoc analysis. Data from human samples were compared using the Student’s t test or Mann-Whitney U test, depending on whether they were normally or non-normally distributed. Discrete patient data are presented as percentages, and comparisons between groups were made with Pearson’s χ2 test. When the assumptions of the χ2 test were not met, a Fisher’s exact test was used. For all analyses a p value of <0.05 was considered statistically significant.
RESULTS
TF levels in pulmonary oedema fluid and plasma
The clinical characteristics of the study patients are summarised in table 1. The TF levels in pulmonary oedema fluid from 54 patients with ALI/ARDS were significantly higher than in 28 patients with hydrostatic pulmonary oedema (fig 1A). In patients with ALI/ARDS, TF levels in pulmonary oedema fluid were more than 100-fold higher than simultaneous plasma samples (median 37 113 (interquartile range (IQR) 14 956–73 525) pg/ml vs 336 (IQR 165–669) pg/ml, p<0.001, fig 1B). The very high levels of TF in oedema fluid compared with simultaneous plasma samples strongly suggest an intra-alveolar source of TF. Higher plasma levels of TF were significantly associated with mortality, fewer ventilator-free days, the presence of disseminated intravascular coagulation and the presence of sepsis in patients with ALI/ARDS. There were no differences in median tidal volume between patients with hydrostatic oedema and those with ARDS (10.4 ml/kg vs 10.3 ml/kg, p = 0.675), or in median positive end expiratory pressure at the time of oedema fluid sampling between those with hydrostatic oedema and those with ARDS (5.0 vs 7.5, p = 0.072). Minute ventilation was significantly higher in patients with ALI/ARDS (hydrostatic 13.5 l/min vs ARDS 15.7 l/min, p = 0.01), which is probably a reflection of increased dead space ventilation in ALI/ARDS.22 When patients with ARDS with direct lung injury (aspiration and pneumonia) were compared with patients with indirect lung injury (sepsis, transfusion reaction, neurogenic oedema), there were no differences in oedema fluid or plasma TF levels, severity of illness score, days of mechanical ventilation, ventilator-free days, ICU days or mortality.
Clinical characteristics of 54 patients with ALI/ARDS and 28 patients with hydrostatic pulmonary oedema from whom plasma and pulmonary oedema fluid were obtained, and 12 patients from whom lung tissue was obtained for immunohistochemistry

Comparison of plasma and pulmonary oedema fluid levels of tissue factor (TF) and clot time in patients with acute lung injury/acute respiratory distress syndrome (ALI/ARDS) and control patients with hydrostatic pulmonary oedema (HYDRO). (A, B) Boxplots of TF protein levels in (A) oedema fluid and (B) plasma measured by ELISA. TF protein levels in pulmonary oedema fluid were significantly higher in ALI/ARDS vs HYDRO (*p = 0.012, Mann-Whitney U test) and TF protein levels in plasma were significantly higher in ALI/ARDS vs HYDRO (**p = 0.02, Mann-Whitney U test). Note that TF levels in oedema fluid (A) are more than 100-fold higher than simultaneous levels in plasma (B) in both patient groups. (C, D) Boxplots of clot time measured by recalcification time of normal plasma mixed with pulmonary oedema fluid from patients with ALI/ARDS or HYDRO in the absence (C) or presence (D) of a TF blocking antibody. Clot time was significantly longer in plasma mixed with oedema fluid from patients with HYDRO vs ALI/ARDS (†p = 0.006, Mann-Whitney U test), and this difference was negated when TF activity was blocked (p = 0.095, Mann-Whitney U test).
Effect of pulmonary oedema fluid on clot formation in normal plasma
Clot time of normal plasma was significantly shorter in the presence of pulmonary oedema fluid from patients with ALI/ARDS than with pulmonary oedema fluid from patients with hydrostatic oedema (fig 1C). This difference was negated by pre-incubation of pulmonary oedema fluid samples with a blocking antibody to TF (fig 1D). TF protein levels were inversely associated with clot time in both ALI/ARDS and hydrostatic pulmonary oedema (ALI/ARDS: r = –0.616, p<0.001; hydrostatic pulmonary oedema: r = –0.757, p<0.001).
TF immunohistochemistry in human lung
Six patients who died from ALI/ARDS and six control patients who died without pulmonary disease were studied. These patients were a different group from those from whom pulmonary oedema fluid and plasma were obtained. Demographic and clinical characteristics of these patients are summarised in table 1. Patients who died with ALI or ARDS tended to be younger, had more organ failures and had a longer duration of assisted ventilation than patients who died without pulmonary disease, although the differences were not statistically significant. The most common causes of ALI or ARDS were sepsis (pulmonary or nonpulmonary, n = 3) and pneumonia without sepsis (n = 1). The diagnoses of the control patients who died of non-pulmonary causes were cardiovascular or central nervous system disease. The immunohistochemical results shown in fig 2 were selected because they are typical for the six patients each who died with ALI/ARDS compared with those who died of non-pulmonary causes. TF immunostaining was displayed by cuboidal alveolar epithelial cells (fig 2A), squamous alveolar epithelial cells (fig 2B), neutrophils and alveolar macrophages in the airspaces, as well as within hyaline membranes in lung tissue sections from patients who died with ALI/ARDS (fig 2A, B). These immunostaining patterns for TF were not seen in tissue sections from patients who died of non-pulmonary causes (fig 2C). Double immunofluorescence staining for TF (green) and pro-SP-B (red), an alveolar epithelial type II cell marker, showed that TF co-localised (yellow) to alveolar epithelial type II cells and alveolar epithelial type I cells in patients who died with ALI/ARDS (fig 2F, G). Some yellow double immunofluorescent staining appears ambiguous with respect to the type of alveolar epithelial cell (fig 2F). This ambiguity appeared to be related to a transition in the cell phenotype from alveolar epithelial type II cells to type I cells during the reparative phase, which is typified by type II cell hyperplasia (fig 2A) that is followed by transition to type I cells as the injured lung tissue is repaired. In other areas along the alveolar membrane, TF was evident alone in alveolar epithelial type I cells (green; fig 2F, G). Minimal TF staining and co-localisation was seen in the lungs of patients who died without ALI/ARDS (fig 2H).

Immunohistochemical localisation of tissue factor (TF) in lung tissue sections from patients who died with acute respiratory distress syndrome ((+) ARDS) or without pulmonary disease ((−) ARDS). (A) Brown reaction product is visible in inflammatory cells (*) in the airspaces and along the epithelial cell lining (arrow) of the airspaces of the lung tissue section from a patient who died with ARDS. Cuboidal alveolar epithelial cells (arrow) are immunostained brown, indicating TF protein localisation. (B) In areas of thickened distal airspace walls of another patient, the squamous alveolar epithelial cells are distinctly immunostained brown (arrow). In contrast, immunolabelling of the occasional inflammatory cells in the airspaces is faint and patchy, and immunolabelling of alveolar epithelial cells is absent in the lung tissue from a patient who died of non-pulmonary disease (C). Immunostaining controls (omission of the primary antibody) are negative for lung tissue sections from a patient who died with ARDS (D) or with non-pulmonary disease (E). (F, G) Double immunofluorescence staining was used to identify the cell type of alveolar epithelial cells with TF immunostaining. TF protein was immunolabelled with green fluorochrome and pro-SP-B was immunolabelled with red fluorochrome. Squamous alveolar epithelial cells are immunostained green in the tissue sections from the patients who died with ALI/ARDS (large arrows), and cuboidal alveolar epithelial cells are immunostained red (pro-SP-B) or yellow (co-localisation of TF and pro-SP-B; short arrows) in the same tissue sections. (H) By comparison, only cuboidal alveolar epithelial cells are immunostained red (pro-SP-B) or yellow (co-localisation of TF and pro-SP-B; short arrows) in the tissue section from a patient who died from non-pulmonary disease. All panels are the same magnification (scale bars in E and H = 100 μm).
TF activity on A549 cells
To determine whether alveolar epithelial cells can express active TF, TF activity was measured in monolayers of A549 cells. Under basal conditions, A549 cells had almost no measurable TF activity (fig 3A, C). However, TF activity was upregulated in a dose-dependent fashion when the cells were exposed to increasing amounts of cytomix (fig 3A). As little as 1 ng/ml of each cytokine resulted in a significant increase in TF activity on the cell surface. There was also a time-dependent increase in TF activity when A549 cells were exposed to 20 ng/ml cytomix for increasing lengths of time (fig 3C). TF activity did not increase significantly until the 6 h time point and peaked at 16 h before declining slightly at 24 h (p<0.05 vs control, one-way ANOVA with post hoc Tukey test).

Tissue factor (TF) activity in A549 cells increased in a dose-dependent (A) and time-dependent (C) manner in response to incubation with cytomix (tumour necrosis factor α, interleukin 1β, interferon γ). TF protein concentration, measured by ELISA, also increased in a dose-dependent (B) and time-dependent (D) manner in cell lysates under the same conditions (*p<0.05 vs controls in all experiments, one-way ANOVA with post hoc Tukey test). Data presented as mean (SD).
TF protein concentrations in A549 cells
There was a time-dependent and dose-dependent increase in TF protein levels as measured by ELISA in cell lysates from A549 cells exposed to cytomix (fig 3B, D) that paralleled the increase in TF activity (p<0.05 vs control, one-way ANOVA with post hoc Tukey test). The rise in protein concentration in the cell lysates peaked at 6 h.
TF gene expression
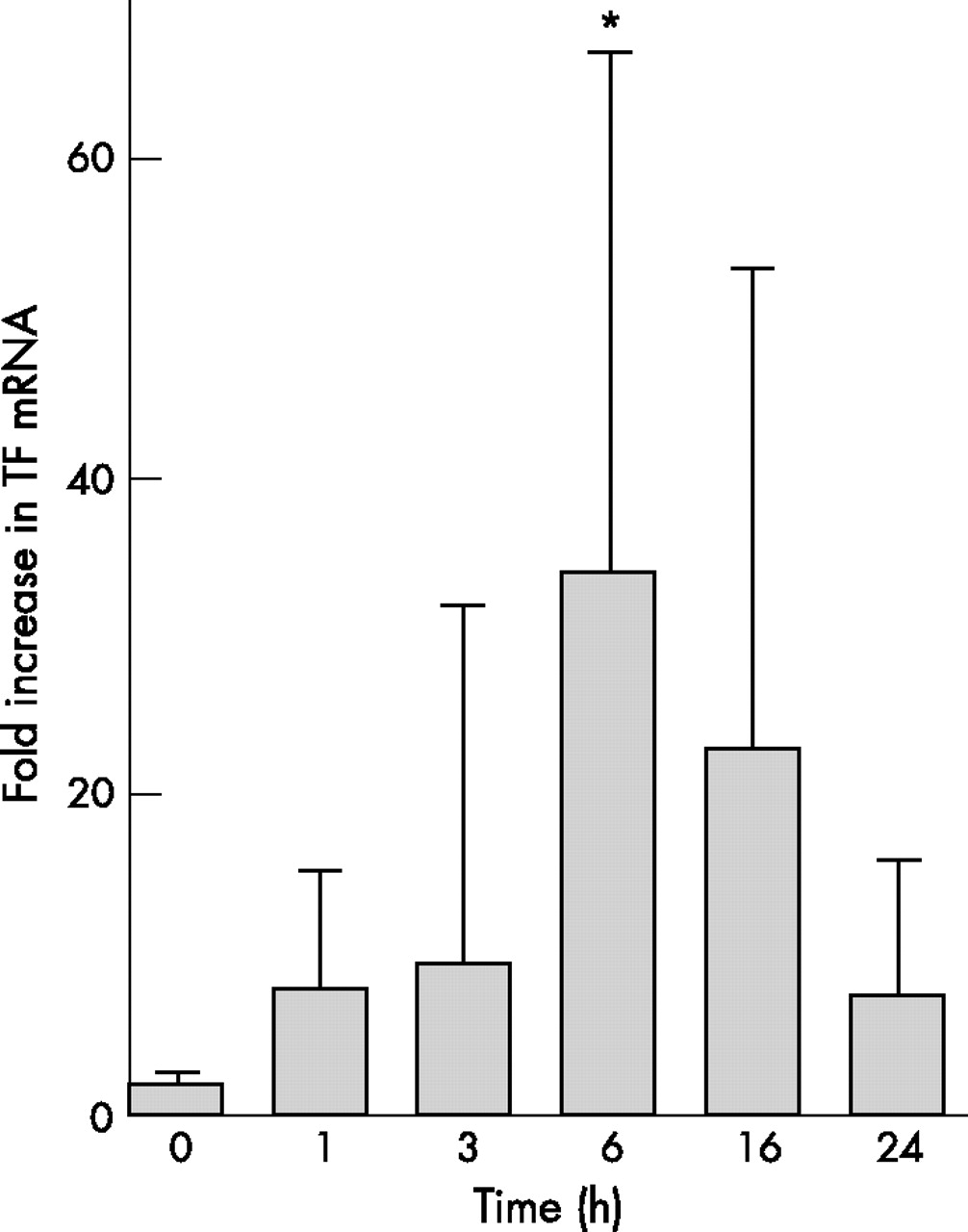
TF mRNA was not present in A549 cells at baseline but was upregulated in a dose-dependent and time-dependent manner after exposure to cytomix compared with β-actin (p = 0.006 vs 0 h, p = 0.025 vs 1 h, p = 0.037 vs 3 h, p = 0.024 vs 24 h, one-way ANOVA with post hoc Tukey test for multiple comparisons, fig 4). When cells were treated with 5 μg/ml actinomycin D in addition to 20 ng/ml cytomix, there was no increase in TF mRNA from 0 to 6 h, which suggests that changes in TF mRNA with cytomix stimulation are a result of new gene transcription and not secondary to changes in mRNA stability (data not shown).

Increase in tissue factor (TF) gene expression in response to cytomix. TF mRNA was measured using semi-quantitative PCR and fold-change in mRNA was calculated using the 2ΔΔCt method. When exposed to 20 ng cytomix for increasing lengths of time, TF mRNA increased 34-fold at 6 h and was still increased above baseline (time 0) at 16 h. *p = 0.006 vs 0 h, p = 0.025 vs 1 h, p = 0.037 vs 3 h, p = 0.024 vs 24 h by one-way ANOVA with post hoc Tukey test. Data presented as mean (SD).
Effect of human pulmonary oedema fluid on TF activity in A549 cells
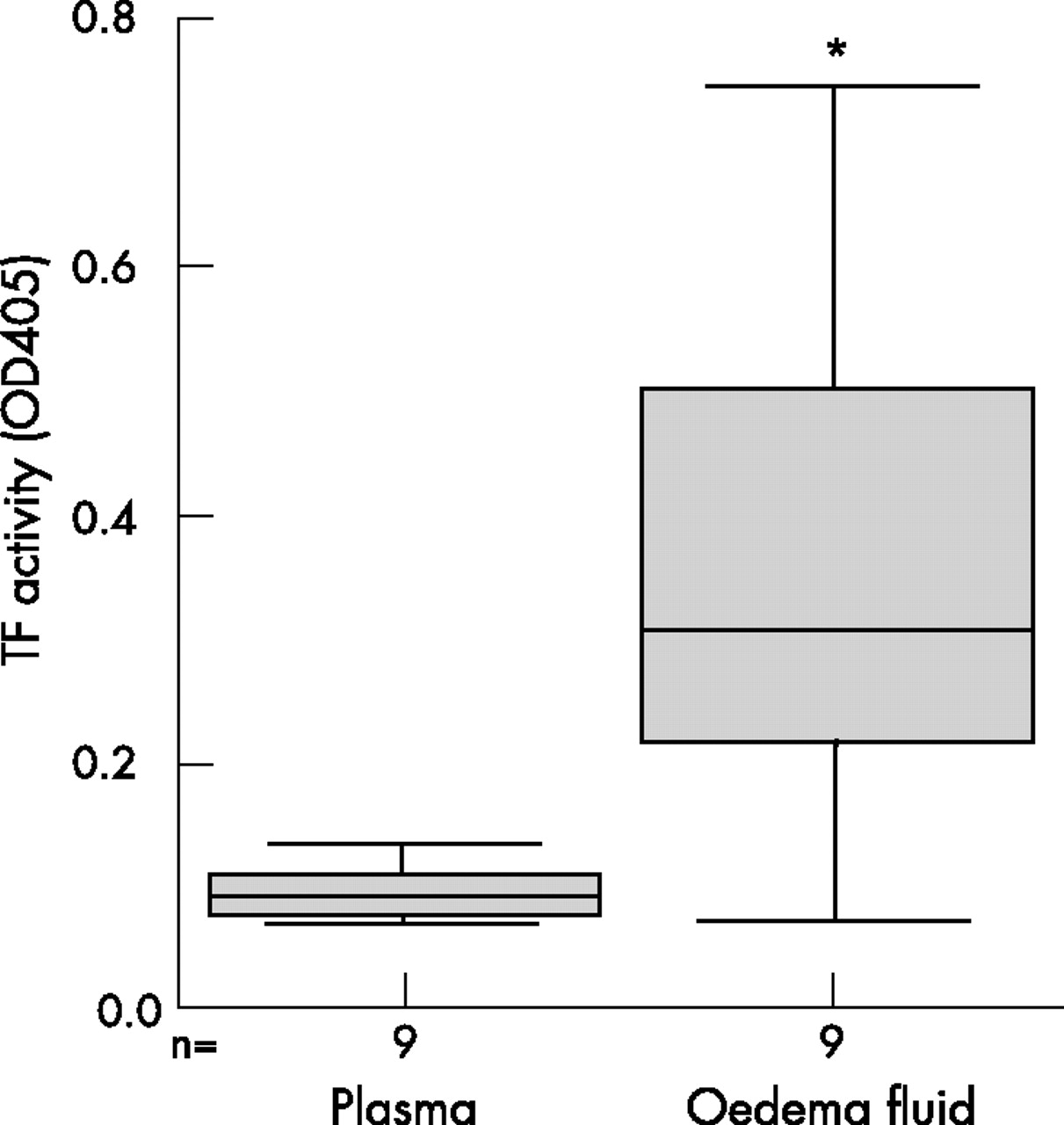
To determine the effect of a physiological stimulus on TF activity in A549 cells, cells were exposed to pulmonary oedema fluid or plasma from patients with ALI/ARDS. TF activity was increased on A549 cells following incubation with human pulmonary oedema fluid from patients with ARDS, but not following incubation with plasma samples from the same patients (p = 0.003 vs plasma, Mann-Whitney U test, fig 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Boxplot comparing tissue factor (TF) activity on A549 cells following exposure to pulmonary oedema fluid or plasma from patients with acute lung injury/acute respiratory distress syndrome (ALI/ARDS). Generation of factor Xa by A549 cells was measured with a chromogenic substrate and expressed as OD405 following incubation with oedema fluid or plasma samples from nine patients with ALI/ARDS (*p = 0.003, Mann-Whitney U test).
TF in small airway epithelial cells (SAEC)
Primary isolates of human distal SAEC were also assessed for TF gene and protein expression and cell surface TF activity. Under basal conditions in culture, these cells contain TF mRNA and TF protein and have high levels of cell surface TF activity (data not shown), which suggests that they may also have a role in intra-alveolar fibrin deposition in lung injury.
DISCUSSION
TF is the most potent initiator of the extrinsic coagulation cascade.23 The alveolar compartment in patients with ALI/ARDS is a procoagulant antifibrinolytic environment, and intra-alveolar generation of thrombin and fibrin probably has significant proinflammatory effects. We first tested whether TF was present in pulmonary oedema fluid and plasma from patients with ALI/ARDS and compared them with a group of patients with hydrostatic pulmonary oedema. TF protein levels were more than 100-fold higher in pulmonary oedema fluid than in plasma from the same patients. Plasma levels of TF in patients with ALI/ARDS correlated with mortality, ventilator-free days and the presence of sepsis, which suggests that systemic activation of coagulation may be clinically important in ALI. In addition to measuring protein levels, we also measured the ability of pulmonary oedema fluid to stimulate clot formation in normal plasma. Oedema fluid from both patients with hydrostatic oedema and those with ALI/ARDS stimulated clot formation but the time to clot formation was significantly shorter in patients with ALI/ARDS, which confirms that the alveolar compartment in ALI/ARDS is procoagulant. In addition, the difference in clot time between hydrostatic oedema and ALI/ARDS disappeared when TF activity in oedema fluid was inhibited, which suggests that the difference in clot time between hydrostatic oedema and ALI/ARDS can be explained by the activity of TF. There were no differences in tidal volume, positive end expiratory pressure or time to oedema fluid sampling between patients with hydrostatic oedema and those with ALI/ARDS, suggesting that the differences seen were not due to differences in mechanical ventilation.
Previous work has shown that there is an increase in TF-dependent procoagulant activity in the alveolar compartment in ARDS.2,4 However, these studies were performed on BAL fluid which has a variable degree of dilution, making comparison between patients difficult. In addition, TF levels in the alveolar compartment in ARDS have never been compared with an appropriate control group such as patients with acute respiratory failure due to acute hydrostatic pulmonary hydrostatic oedema. In the current study, direct aspiration of undiluted pulmonary oedema fluid allowed a direct comparison of levels and activity of TF in the alveolar compartment with other patients and simultaneous plasma samples, and enabled us to compare these measurements with a control group of critically ill patients with hydrostatic pulmonary oedema.
Given the markedly increased levels of TF in pulmonary oedema fluid compared with plasma, we hypothesised that there is an intra-alveolar source of TF. To determine the origin of intra-alveolar TF, we used immunohistochemistry to localise TF in human lung tissue. The alveolar epithelium stained positively for TF in ARDS but not in control lung tissue, suggesting that it may be a source of intra-alveolar TF in patients with ALI. This is in contrast to previous literature which reported TF staining of the alveolar epithelium in normal human lungs.24,25 However, these earlier studies made only a limited examination of TF staining in the lung as part of studies of TF expression in all tissues; no co-localisation was done to confirm alveolar epithelial expression. Our study is the first to carry out a detailed examination of the location of TF in the lung in patients with ALI/ARDS, together with co-staining of the alveolar epithelium to identify the specific cellular type expressing TF. In vitro studies with A549 cells confirmed that the alveolar epithelium expresses active TF in response to a pro-inflammatory stimulus.
The alveolar epithelium, which covers a large surface area in the alveolar compartment, is known to participate in the modulation of coagulation through the protein C pathway.26 It also has other important functions in ALI such as surfactant production and alveolar fluid clearance27 by type II cells and sodium transport by type I cells,28 suggesting that both cell types have critical roles in the pathogenesis and resolution of ALI. Few data exist regarding TF expression by cultured alveolar epithelial cells. One study reported a low level of TF activity in unstimulated A549 cells.29 Previous studies have shown that rat alveolar epithelial cells in culture produce functionally active TF;13,14 however, as mentioned previously, these studies had contradictory results with one demonstrating a high level of TF activity in isolated alveolar epithelial cells that did not change with an inflammatory stimulus14 and the other showing a decrease in TF activity following an inflammatory stimulus.13 In addition, there is little information on the time course of TF mRNA expression, protein expression and TF-dependent procoagulant activity in the alveolar epithelium. Our findings show that alveolar epithelial cells are capable of producing functional TF on the cell surface when exposed to pro-inflammatory cytokines or to pulmonary oedema fluid aspirated from patients with ALI/ARDS. This finding suggests that the alveolar compartment contains mediators that are potent inducers of TF activity on the alveolar epithelium. The increase in TF activity on A549 cells results from increased TF mRNA transcription, which suggests that, at baseline, the alveolar epithelium may not be capable of initiating coagulation but becomes procoagulant following exposure to an inflammatory stimulus.
Our study is unique in that it provides detailed information about the time course of TF mRNA and protein production in alveolar epithelial cells and correlates this with cell surface Xa generating activity, findings that have not been previously reported. In addition, the ability of the airway epithelium to modulate intra-alveolar coagulation through active TF expression was confirmed in primary SAEC that contain TF mRNA and active cell surface TF basally. SAEC also play an important role in the extensive apoptosis seen in ALI.30 In normal lung tissue taken at necropsy, low levels of TF were found in the alveolar epithelium; however, there was a dramatic increase in TF in the lungs of patients with ALI/ARDS. The TF was localised to alveolar epithelial cells, both type I and type II cells, as well as to macrophages and hyaline membranes.
Several experimental animal studies have shown that TF expression in the lung is low at baseline but increases in response to systemic administration of lipopolysaccharide,31,32 hypoxia33 or bleomycin lung injury.34 TF mRNA expression is increased in the lung and localises to the alveolar epithelium in a rabbit model of endotoxaemia.35 Our study shows that alveolar epithelial TF expression increases in response to an inflammatory stimulus, a mechanism that may be important in the pathogenesis of ALI. In a lethal baboon model of E coli sepsis, systemic blockade of TF resulted in attenuated lung injury measured by physiological parameters and histology,8,9 highlighting the important role of TF in the development of lung injury. We have demonstrated TF immunostaining in alveolar epithelial cells and hyaline membranes in lung tissue from patients with ALI/ARDS, further supporting the hypothesis that the alveolar epithelium upregulates TF in response to inflammatory stimuli in the setting of ALI/ARDS and may be important in the pathogenesis of ALI/ARDS.
Several questions remain unanswered. While the high levels of TF found in human oedema fluid compared with plasma are striking, the exact mechanism by which the TF is released into the alveolar compartment is unknown. It is clear that there is significant injury to and sloughing of the alveolar epithelium in patients with ALI/ARDS.36 Necrotic cells and apoptotic cells are a potential source for the very high intra-alveolar levels of TF in our patients. Alternatively, the TF could be cleaved from the cell surface or actively secreted. TF may also be released from alveolar macrophages.
There are some limitations to this study. First, we studied A549 cells, a human cancer cell line which may not accurately represent in vivo responses, although these cells have been used in many cell and molecular investigations. However, the ability of the alveolar epithelium to express active TF was confirmed in primary isolates of SAEC which have TF mRNA and functional protein basally. In addition, the demonstration of TF in the alveolar epithelium in human lung tissue from patients with ALI/ARDS gives credence to our in vitro experiments. Second, while our in vitro studies characterise the expression of TF in the alveolar epithelium, they do not explore the functional consequences of TF expression in ALI/ARDS. The ultimate consequences of this intense procoagulant stimulus in the alveolar compartment in patients with ALI/ARDS are unknown. There are several parallels between the intense tissue injury and coagulation abnormalities in the lung in ALI/ARDS and the systemic microvascular injury and thrombosis that develop in severe sepsis. Coagulation initiation and thrombus formation are central to the pathogenesis of sepsis,37 and modulation of coagulation with activated protein C can correct these derangements and improve patient outcome.5 It is possible that alveolar coagulation is similarly detrimental, and production of thrombin, fibrin and other coagulation proteins may promote injury and inflammation. Conversely, alveolar fibrin deposition may be important in localising inflammation and preserving alveolar architecture in the acute phase when there is significant necrosis of the alveolar epithelium.
The current findings are important for several reasons. First, they further substantiate the alveolar epithelium as a source of intra-alveolar coagulation proteins that may be critical to the pathogenesis of ALI/ARDS and provide the first detailed immunohistochemical localisation of TF in the alveolar compartment in ALI/ARDS. Second, by focusing on TF, we have examined the initial steps of intra-alveolar fibrin deposition. By understanding the first critical steps that lead to intra-alveolar fibrin deposition and hyaline membrane formation, we may be better able to modulate these end-points compared with modulating other coagulation proteins in the coagulation cascade. Finally, by identifying the alveolar epithelium as a significant source of coagulation proteins and characterising in detail the increase in production of TF by an inflammatory stimulus, we are paving the way for epithelial-directed treatment that could be delivered by inhalation, thereby avoiding the potential deleterious effects of systemic treatment and specifically targeting a key pathogenic mechanism in the development of ALI/ARDS.
Acknowledgments
The authors thank Nancy Wickersham and Nimet Browne for their technical assistance.
REFERENCES
Footnotes
-
Published Online First 13 March 2007
-
Supported by National Institutes of Health grant HL70521 and HL081332 to LBW and HL51856 and HL74005 to MAM, an American Lung Association Biomedical Research Grant to LBW, and the Vanderbilt Physician Scientist Development Award to JAB.
-
Competing interests: None.