Article Text

Abstract
Background: This study investigated the addition of salmeterol to existing treatment for exacerbations in patients with poorly reversible chronic obstructive pulmonary disease (COPD).
Methods: 634 patients aged >40 years with a history of COPD exacerbations (including at least two in the previous year) and poor reversibility of airflow obstruction (⩽10% predicted forced expiratory volume in 1 second) received either salmeterol 50 μg or placebo twice daily from a Diskus inhaler for 12 months. The primary outcome was the number of moderate and severe exacerbations.
Results: The median rate of moderate or severe exacerbations in the intent-to-treat (ITT) population was lower in the salmeterol group (0.00, range 0.0–9.8, n = 316) than in the placebo group (0.93, range 0.0–13.0, n = 318), but the difference was not statistically significant (p = 0.27). The median rate of exacerbations in the per protocol population (>90% compliance) was also found to be lower in the salmeterol group (0.00, range 0.0–5.0, n = 206) than in the placebo group (0.93, range 0.0–5.6, n = 195) and did reach statistical significance (p = 0.007). For secondary end points, patients receiving salmeterol had significant improvement in lung hyperinflation measured by inspiratory capacity which was evident at 4 weeks and maintained over 12 months (p = 0.035), and a significant improvement in health status measured by the St George’s Respiratory Questionnaire at 12 months (p = 0.002).
Conclusion: Salmeterol has a positive effect on symptoms and health status of patients with COPD when added to usual treatment. Exacerbations are only reduced in patients who comply with treatment.
- COPD, chronic obstructive pulmonary disease
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- IC, inspiratory capacity
- ICS, inhaled corticosteroid
- ITT, intent-to-treat
- LABA, long acting β agonist
- MMEF, maximum mid-expiratory flow
- PPP, per protocol population
- SGRQ, St George’s Respiratory Questionnaire
- SVC, slow vital capacity
- chronic obstructive pulmonary disease
- exacerbations
- inspiratory capacity
- salmeterol
Statistics from Altmetric.com
- COPD, chronic obstructive pulmonary disease
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- IC, inspiratory capacity
- ICS, inhaled corticosteroid
- ITT, intent-to-treat
- LABA, long acting β agonist
- MMEF, maximum mid-expiratory flow
- PPP, per protocol population
- SGRQ, St George’s Respiratory Questionnaire
- SVC, slow vital capacity
Chronic obstructive pulmonary disease (COPD) is characterised by progressive airflow limitation that is not fully reversible and is a major cause of chronic morbidity and mortality throughout the world.1
Patients with moderate and severe COPD are particularly prone to exacerbations, which increase in frequency with the severity of disease2 and may have a considerable impact on the quality of life of patients.3 Although long term evaluation of the disease has been based historically on the rate of decline in lung function over time, recently more emphasis has been focused on the frequency and severity of exacerbations.4 Several studies have shown that long acting β2 agonists (LABAs) such as salmeterol and formoterol are beneficial in COPD, but most of these studies have been relatively short and in groups of homogeneous patients with careful control of other medications.5–8 We have therefore investigated the effect of salmeterol on the rate of moderate to severe exacerbations over one year in a “real life” setting in which COPD patients with poorly reversible airflow obstruction were chosen (a priori) to be treated with salmeterol or placebo, in addition to their concurrent treatment. Since lung hyperinflation due to air trapping is commonly observed in these patients, the effect of salmeterol on this parameter was also assessed by measuring the inspiratory capacity, in addition to other indicative parameters of lung function, breathlessness and exercise capacity, symptoms and change in health status.
METHODS
Subjects
Patients aged ⩾40 years with COPD according to the ERS consensus statement9 were recruited from hospital respiratory units in 19 European countries. All were current or former smokers with a baseline pre-bronchodilator forced expiratory volume in 1 second (FEV1) of <70% predicted10 and poor reversibility (defined as an increase in FEV1 of ⩽10% predicted in response to 400 μg inhaled salbutamol). Patients were required to have an established history of COPD exacerbations, including at least two in the previous year that had required treatment with antibiotics and/or oral corticosteroids. Those who required regular oxygen therapy or had received any change in COPD medication in the 4 weeks before the study were excluded. Doses up to 2000 μg/day beclomethasone dipropionate or equivalent or 1000 μg/day fluticasone propionate were allowed during the study, as were usual treatments such as methylxanthines, short acting anticholinergic agents, and mucolytics. Long acting anticholinergic agents were not available at the time the study was conducted.
The study protocol was approved by the investigational centre research ethics committees and conducted according to Good Clinical Practice guidelines and the 1996 Declaration of Helsinki. Signed informed consent was obtained from all patients before enrolment.
Study design
This randomised double blind study was conducted between April 2000 and November 2001. Eligible patients entered a 2 week run-in period and all short acting β2 agonists were replaced with salbutamol (100 μg/puff) for symptomatic relief and any LABAs were discontinued. Patients who completed the run-in and were stable on their COPD medication were then randomised to receive either salmeterol 50 μg or identical placebo twice daily administered from a Diskus inhaler for 12 months. Patients were assigned using a computer generated randomisation schedule (Patient Allocation for Clinical Trials, GlaxoWellcome, UK) stratified for smoking history and inhaled corticosteroid (ICS) use at entry (block size of four), and centrally randomised using an Interactive Voice Recognition System. Patients attended the clinic at weeks 4, 16, 28, 40 and 52 during the treatment period and within 2 days of the onset of any exacerbation. Such episodes were followed up by telephone contact or visits at 7 day intervals until resolution.
Measurements
The primary end point in the study was the number of exacerbations in the intent-to-treat (ITT) population with secondary end points of mild exacerbations, lung function, diary card parameters, and health status. Exacerbations were identified using an event based definition in which a worsening of symptoms required a change in medication. Exacerbations were classified as moderate if they required treatment with antibiotics and/or oral corticosteroids or an increase in ICS dose, and severe if they required admission to hospital. Mild exacerbations were defined as worsening symptoms managed with increased use of salbutamol alone. This classification is consistent with that used widely in recent clinical trials.11
Spirometric measurements were made at each clinic visit. The highest of three measurements of FEV1, forced vital capacity (FVC) and, at some centres where facilities permitted, the slow vital capacity (SVC), maximum mid-expiratory flow (MMEF), and inspiratory capacity (IC) (defined as the difference between total lung capacity and functional residual capacity) were recorded. Wherever possible, spirometric tests were performed at the same time of day as the randomisation visit using the same spirometer (always calibrated on each study day in accordance with the manufacturer’s specification) and supervised by the same study staff. All measurements were made at least 12 hours after the study medication, and relief salbutamol was withheld for 4–6 hours before measurement.
Assessment of the general perception of breathlessness was made using the Medical Research Council (MRC) dyspnoea scale. At centres able to perform an incremental shuttle walk exercise test,12 patients were asked to indicate their perception of breathlessness before and after completing the test using the Borg CR-10 scale.13
Health status was assessed at baseline and at all clinic visits using the disease specific St George’s Respiratory Questionnaire (SGRQ)14 in 14 countries where the questionnaire had validated translations. Patients were asked to record morning peak expiratory flow rate (PEF) before taking COPD medication, the number of occasions they had used salbutamol for relief of symptoms, the severity of symptoms including cough, sputum consistency, volume and colour, breathlessness and night-time awakenings due to COPD, and any changes in medication on the daily record card. Compliance was assessed by recording the number of remaining doses on the integral counter of the Diskus inhalers and comparing it with the number that should have been left if the patient had taken all their medication. Safety was assessed by adverse event monitoring.
Statistical analysis
The sample size was sufficient to detect a difference of 0.5 exacerbations a year at the 5% significance level with 90% power, assuming a standard deviation of 1.7 exacerbations, and adjusting for the asymptomatic relative efficiency of the Wilcoxon-Mann-Whitney test.
Data for patients who withdrew from the study were included up to the point of withdrawal, unless otherwise stated. The number of moderate and severe exacerbations experienced during the study was imputed for those patients without complete data (early withdrawal) to provide an estimate of the number of exacerbations over a 52 week period. Imputation was based on 4 weekly data. The number of exacerbations in a year was calculated by multiplying the number of exacerbations the patient had by 13 and dividing by the number of 4 weeks the patient was in the study. The numbers of mild, moderate, and severe exacerbations in both the ITT and per protocol populations (PPP; defined as ITT group without major protocol violaters) were analysed using the Wilcoxon-Mann-Whitney rank sum test with the van Elteren extension15 after stratification for ICS use, smoking status, and use of LABAs before the trial. Because of the nature of the data, there were a number of tied values. For the purpose of the analysis the ranks for the tied data were averaged and the average score assigned to each tied observation. Major violations (defined before analysis) included patients whose compliance with study medication was <90%, those who received treatment for <50 weeks, those who used prohibited medication during the study, and those not fulfilling the entry criteria.
Clinic pre-bronchodilator FEV1 and morning PEF were analysed using analysis of covariance. No data were carried forward for premature withdrawals. Smoking status, use of LABAs before the trial, and ICS use were covariates in the model, along with other treatment. In addition, for morning PEF the baseline value, age and sex were also included in the model. For reliever use, the median was calculated for each patient for the run-in period at baseline and for the treatment period using all available data. For other data measured on the daily record card, mean values were calculated for baseline and the treatment period, and the significance in the change from baseline was analysed using the Wilcoxon-Mann-Whitney rank sum test or the two sample t test if the data were normally distributed. The MRC dyspnoea score data were analysed during the treatment period using logistic regression for ordinal data. The shuttle walk test was analysed using the Wilcoxon-Mann-Whitney rank sum test. For the SGRQ, a transformed score was calculated for each of the three sub-scales of the questionnaire (symptoms, impacts and activity) and the overall total score in accordance with the developers’ scoring guidelines. If patients withdrew early, the last observation was carried forward for the analysis. Treatment comparisons for change from baseline were assessed using analysis of covariance with age, sex, ICS use, smoking status, and baseline scores as covariates. These comparisons were carried out for the total score and for each of the three domains for the SGRQ.
RESULTS
Of the 726 patients recruited, 634 were randomised; 316 received salmeterol and 318 received placebo (fig 1). After randomisation, 56 patients (18%) in the salmeterol group and 75 (24%) in the placebo group were withdrawn, mainly due to an adverse event. 233 patients were judged to be protocol violators before unblinding treatment allocation and were omitted to form the PPP group (fig 1). The baseline characteristics of the ITT group are shown in table 1 and indicate a good match between treatment groups. The number of patients continuing other COPD medications (including ICS) was similar between the treatment groups (table 1). During the 12 month study, eight patients (4%) had a change in dose of ICS in the placebo group compared with three (2%) in the salmeterol group. The baseline characteristics of the PPP were also well matched at baseline between treatment groups (data not shown).
Baseline characteristics of randomised population (ITT)

Flow of patients through the study.
Exacerbations
The median number of moderate/severe COPD exacerbations over 12 months in the ITT population was lower in the salmeterol group than in the placebo group. The difference between the groups did not achieve statistical significance (p = 0.27), although there was a 21% reduction in the mean rate with salmeterol (1.18 v 0.93 per patient per year). However, in the PPP, there were significantly fewer (p = 0.007) moderate/severe exacerbations on salmeterol than on placebo (table 2), with a mean rate of reduction of 30% (0.83 v 0.58). No significant effect of ICS, anticholinergic agents, or xanthine use was seen on the median reduction in the rate of moderate/severe exacerbations in the ITT population for salmeterol compared with placebo. Overall, the majority of subjects (79%) did not use a LABA prior to the study and had a similar number of exacerbations in each group (table 2). For subjects who used a LABA before the study, a 48% reduction was seen in the salmeterol group (0.99 exacerbations) compared with placebo (1.91 exacerbations, table 2).
Number of moderate/severe COPD exacerbations
The number of patients reporting at least one mild exacerbation was similar in the groups (19% placebo; 20% salmeterol) and the overall mean (SD) rate was 0.44 (1.19) for the salmeterol group and 0.38 (1.0) for the placebo group (p = 0.92).
Lung function/volume
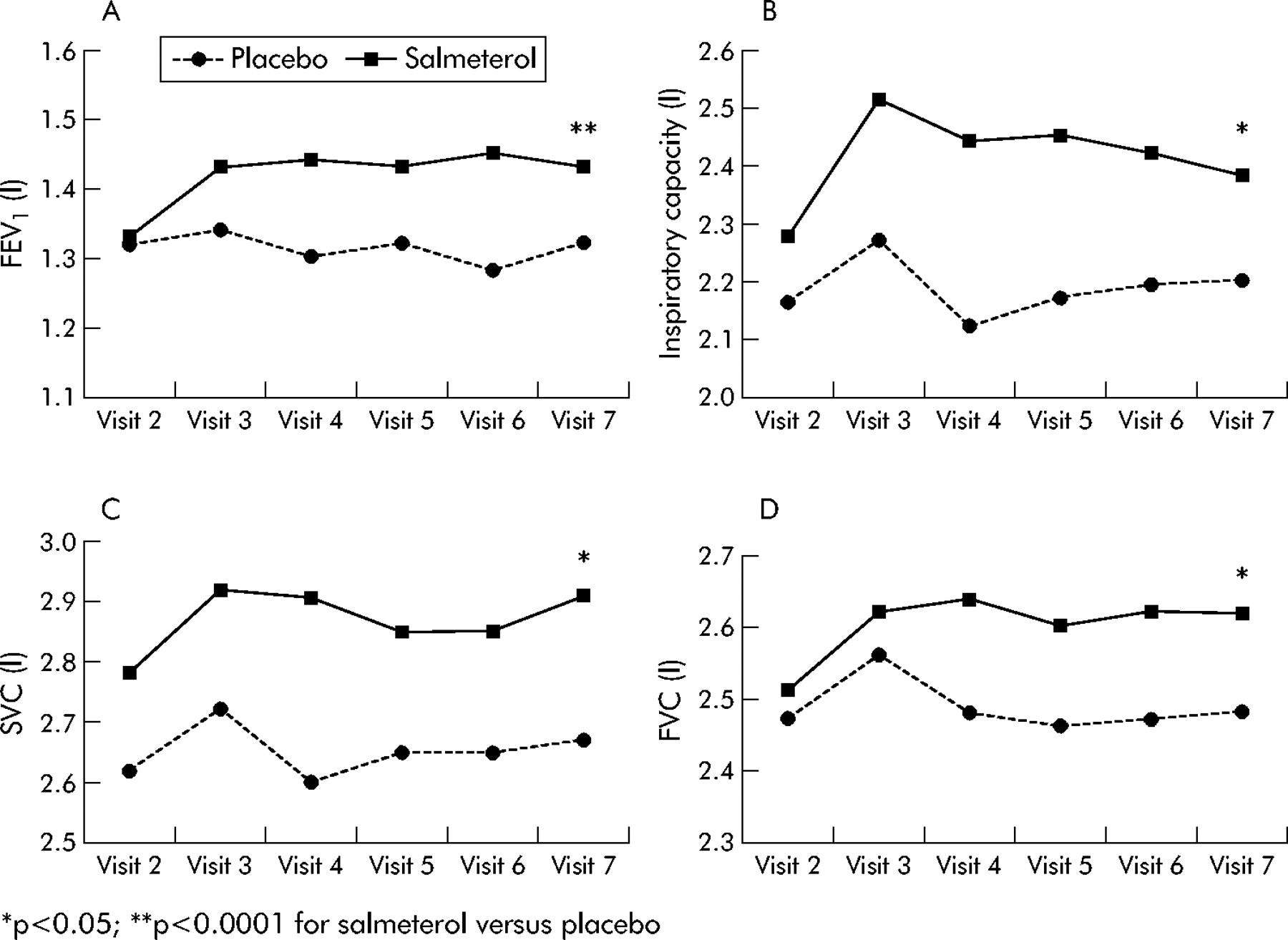
Salmeterol significantly improved lung function over the 12 month treatment period compared with placebo, with no evidence of tolerance. The increase in IC was evident as early as 4 weeks after starting treatment and was maintained over the 12 month period (fig 2, table 3). An increase in FEV1 was also seen in the salmeterol group compared with placebo from week 4. An improvement in FVC, SVC, IC, and MMEF was also observed in the salmeterol group compared with placebo. The significance of these changes is shown in fig 2 and table 3 at visit 7 (week 52).
Difference between effect of salmeterol and placebo on lung function and volume parameters after 12 months of treatment

Pre-bronchodilator forced expiratory volume in 1 second (FEV1), inspiratory capacity (IC), slow vital capacity (SVC), and forced vital capacity (FVC) over 12 months. Each data point is the mean value for the group (the number of patients evaluated in the salmeterol group and placebo group at visit 2 (baseline) for FEV1 = 316 and 318; FVC = 315 and 318; IC = 152 and 170; SVC = 162 and 177).
Dyspnoea and exercise capacity
At each visit the dyspnoea score using the MRC scale favoured the use of salmeterol, reaching significance at both week 40 (p = 0.019; OR salmeterol/placebo = 1.47) and week 52 (p = 0.029; OR = 1.41), and patients receiving salmeterol (n = 219) walked further in the shuttle walk test (30 metres difference at week 52, p = 0.04; 95% CI 0 to 60) than those receiving placebo (n = 207). The perception of breathlessness measured using the Borg scale before and after the shuttle walk was not statistically different between the groups.
Diary card data
There was a significant reduction in the use of relief salbutamol (p<0.0001) and an increase in the proportion of reliever free days (p = 0.009) in the salmeterol group compared with placebo (table 4). Salmeterol resulted in a significant improvement in breathlessness (p = 0.002) and night time awakenings (p = 0.008) compared with placebo. During treatment the mean morning PEF was higher in the salmeterol group than in the placebo group, with a mean difference of 14.61 l/min (95% CI 9.32 to 19.89; p<0.0001). Salmeterol had no effect on sputum production or cough.
Change from baseline in diary card measurements
Health status
At baseline the total SGRQ score was comparable between the groups (49.8 for placebo; 48.3 for salmeterol). Patients receiving salmeterol experienced an improvement at 4 weeks which was maintained throughout the 12 months and was statistically significant at the study end point (fig 3). The change in total score from baseline to 12 months was also significantly better with salmeterol than with placebo (mean (SD) −2.3 (1.10) points on placebo, −6.6 (1.13) on salmeterol; treatment difference p = 0.002). The decrease in scores from baseline to 12 months was also greater for salmeterol than placebo for each of the domains (fig 3).

{kind=link}
{kind=link}
{kind=link}
Mean change from baseline in SGRQ total score and domain scores for (A) salmeterol and (B) placebo at weeks 4, 28 and 52. A lower score indicates an improvement in health status. *p<0.05 compared with placebo.
Safety
Reported events were similar between the treatments. There were no differences in the proportion of patients with serious adverse events, in those withdrawing for adverse events, or in deaths (table 5).
Adverse event profile
DISCUSSION
This study is unique in assessing the effect of the addition of salmeterol to existing treatment, which could include an ICS, in a population of poorly reversible patients, particularly because the patients are investigated in a “real life” setting over a long period of 12 months. Analysis of the primary end point—the number of moderate/severe exacerbations—showed that, although treatment with salmeterol was associated with a 21% reduction compared with placebo in the ITT population, this failed to reach significance. However, a similar reduction in exacerbations has been observed in studies with other long acting bronchodilators such as tiotropium,16 In addition, one year trials with formoterol, using the same definition of exacerbations as the current study, have not shown such a reduction in exacerbations,17,18 which suggests that the reduction in the current study is not a feature of all LABAs.
While it is possible that the lack of significance between the active and placebo treatment groups may be due to the low frequency of exacerbations experienced by the patients, the reasons underlying the lower than expected rates of exacerbations observed in the present study are not clear, particularly in view of the fact that only patients experiencing at least two exacerbations in the previous year were recruited. It is possible that patients who had experienced a maximum of two exacerbations of COPD in the previous year may have been less prone to exacerbations than patients who had experienced a greater number of exacerbations. It is also possible that the placebo effect may have been exaggerated in these patients by merely entering a clinical trial with close monitoring, or that the severity of COPD in the patients recruited was milder than anticipated.
A lower than expected exacerbation rate, however, is not unique in COPD exacerbation studies, and similar issues arose in the study reported by Calverley et al.19 Additionally, we used the same event based definition of exacerbations, which has been endorsed as the most unambiguous and practical approach to clearly defining episodes,11 and has been used in other recently reported long term studies.17,18 The reduction in exacerbations observed in recent studies clearly needs to be taken into account in the design of future intervention studies on exacerbations. A more symptom related definition should perhaps have been used, or more rigorous physiological impairment criteria may need to be used to recruit patients to ensure a more consistent incidence of exacerbations. Further studies are needed to clarify these issues.
Removal of patients deemed to be protocol violators resulted in a higher overall incidence of moderate/severe exacerbations in patients on placebo, although this was still less than two per year. Two of the main criteria for classifying this population (determined before analysis) were 90% compliance with study medication (measured by the dose counter on the Diskus) and/or that they completed at least 50 weeks of treatment. The latter was chosen as it emphasises any benefit of high compliance. If the threshold for compliance in the per protocol population was lowered to 80% (post hoc analysis), there was no significant difference between salmeterol and placebo (p = 0.062). This suggests that a reduction in exacerbations in the salmeterol group was clearly influenced by compliance, and highlights the need for patients to keep taking their medication to obtain a benefit related to the frequency of exacerbations.
The dose of ICS taken by patients during the study was similar between the groups and is therefore unlikely to account for the overall difference in exacerbation rate seen between salmeterol and placebo in these patients. In addition, the number of patients who changed their dose of ICS during the study was small and balanced between the groups, and is also unlikely to have influenced the results. Investigation into the role of prior ICS use on exacerbations showed that, although the addition of salmeterol reduced moderate/severe exacerbations by 27%, the difference in exacerbation rates between ICS or non-ICS users was not significant. Similarly, while a numerical trend towards a reduction in the rate of exacerbations was observed with salmeterol for patients receiving short acting anticholinergic agents, this was not statistically significant. Although previous data have shown that administration of both a LABA and a short acting anticholinergic is more beneficial than either drug alone,6,20,21 our study was not powered to show a difference in these sub-groups of ICS and anticholinergic users and non-users. At the time of the current study, the long acting anticholinergic tiotropium was not available and further research is therefore required to determine whether a LABA and tiotropium have additive effects in COPD, as seen with other combinations of treatment.16–18,22
In COPD, expiratory airflow limitation—the pivotal pathophysiological abnormality—is traditionally measured by FEV1. Salmeterol has been shown to improve FEV1 after 4 weeks,6 as in the current study. An improvement of >100 ml, which was maintained over the 12 months and was greater than placebo at all time points, is likely to be of clinical benefit in patients with a poor baseline lung function and poor reversibility and would be supported by the improvement in health status (particularly the activity domain) observed in the present study. There was no evidence of tolerance to the bronchodilator effects of salmeterol in this long term trial, which confirms the findings of shorter studies.5–7 FVC also showed a similar improvement on salmeterol. However, improvement in both FEV1 and FVC is poorly predictive of dyspnoea and exercise tolerance.23 On the other hand, spirometric IC, which indirectly reflects the end-expiratory lung volume and exercise capacity, can be diminished even at rest in COPD24,25 and provides an indirect measure of hyperinflation which may contribute complementary information to the FEV1 in therapeutic evaluation.23 In the current study salmeterol improved IC within 4 weeks, which was reflected by the reduction in breathlessness and symptoms, as well as improvement in exercise capacity measured by the shuttle walk test. Furthermore, patients receiving salmeterol had a significantly higher percentage of days when they did not use their short acting β2 agonist for relief of symptoms, which may also reflect this improvement in dyspnoea and exercise capacity.
Health status is an established outcome measure for evaluating the efficacy of therapeutic interventions in COPD.26 It has been suggested that exacerbation frequency is an important determinant of health status in COPD and hence an important outcome measure.2 A significant decrease of four units in the SGRQ was seen in patients receiving salmeterol over 52 weeks. Health status was improved in our study as early as 4 weeks after treatment began, and was not only maintained but increased throughout the treatment period—consistent with some previous results27 but greater than in other studies with salmeterol.16,20 Some of these differences may reflect the “real life” situation as patients were allowed to continue with ICS in the current study while in other studies ICS were not permitted.16 Clearly, comparisons between studies need to take into account permitted treatment as well as other inclusion and exclusion criteria.
This study confirms that salmeterol is an effective treatment for patients with COPD. The addition of salmeterol significantly improved lung function (including reduced lung hyperinflation) and dyspnoea, increased exercise capacity, and resulted in an enhanced quality of life. A modest reduction in exacerbations was also seen, but only in patients who showed good compliance with study medication.
Acknowledgments
The authors thank Diana Jones for assistance with the manuscript and Seema Sondhi for assistance in the analysis of health status data.
REFERENCES
Footnotes
-
This manuscript was prepared on behalf of an international study group from 19 countries: G Kaik, N Vetter (Austria); D Popov, D Osmanliev, K Kissiova, P Mandulova, J J Ivanov (Bulgaria); J B Martinot, D Rozen, R Louis, R Pauwels, I J M Stappaerts (Belgium); N Tudoric, F Pavicic, Z Beg Zec, S Skrinjaric Cincar, I Gudelj (Croatia); J Musil, J Krepelka, A Matulova, R Kasak (Czech Republic); J Korsgaard, F F Madsen, H Harving, J I Jensen, P Garsdal (Denmark); S Lane, T McDonnell (Ireland); T Kipper, L Raudla (Estonia); P Hyvernat, M Legendre, P Zuck, S Beaujot, J P Moreau, E Fournier, P Godard, T Similowski, F Ruff, G Pecastaing, N Roche, B Lebrozec, J Y Jasnot (France); H ten Hoff, A Benedix, J Eller, E Liefring, U Westerhausen, W Zachgo, J Bargon, W Westphal (Germany); H Berendsen, H Sinninghe Damste (Holland); I Herjavecz, K Radich, E Juhász, Z Márk, M Jedlinszki, I Áchim (Hungary); V Silins, J Krams (Latvia); W Droszcz, A Szczeklik, M Slominski, J Kruszewski (Poland); J Sanchís, M Rosales Jaldo, S Bardagi, C Esteban, J Serra, JM Rodriguez (Spain); E Rozborilova, P Kristufek, J Mazal, J Simovicova (Slovak Republic); J Sorli, R Kopriva, S Kajba (Slovenia); Y Yashyna, Y Feschenko (Ukraine); G Basran, P M A Calverley, M Wilkins, B Higgins (UK).
-
Funding for the study was provided by Glaxo Wellcome Research and Development (protocol SMS40026).
-
Competing interests: none declared.