Article Text

Abstract
Background: Chronic airway obstruction is characteristic of cystic fibrosis (CF) but there are few studies of airway smooth muscle remodelling in CF.
Methods: Airway smooth muscle content and mean airway smooth muscle cell size were measured by applying design-based stereology to bronchoscopic biopsy specimens obtained from seven subjects with CF and 15 healthy controls.
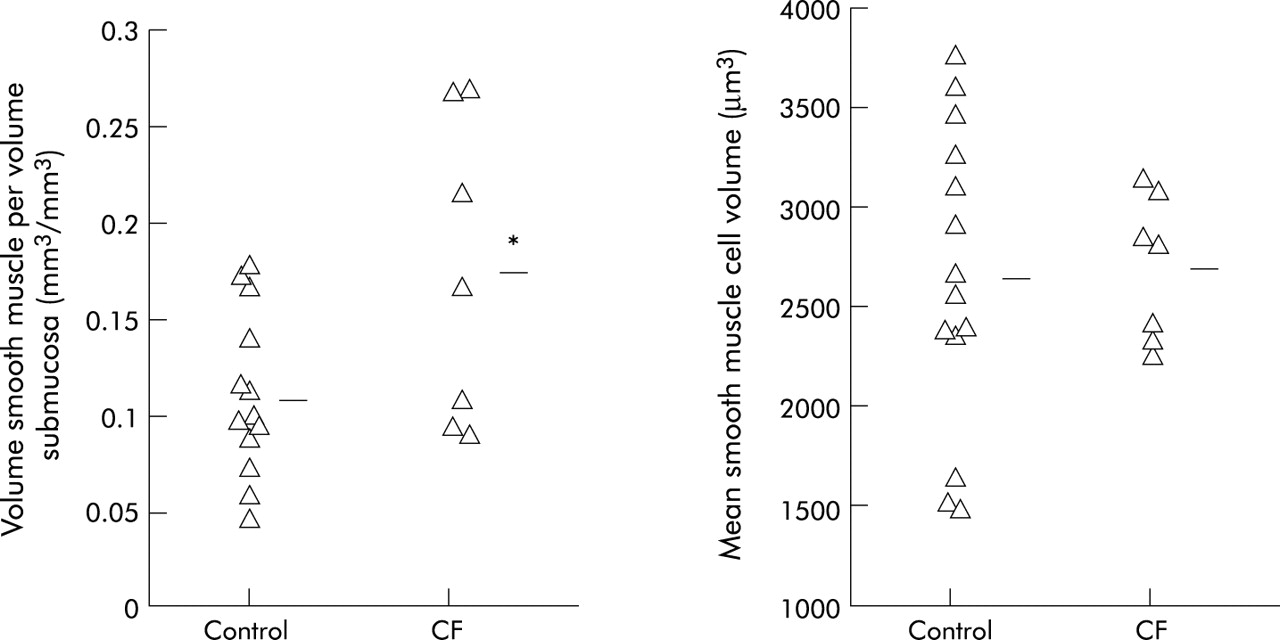
Results: The smooth muscle content increased by 63% in subjects with CF (mean (SD) 0.173 (0.08) v 0.106 (0.042) mm3 smooth muscle/mm3 submucosa, mean difference −0.067; 95% CI −0.12 to −0.013, p = 0.017) but there was no increase in mean cell size (2705 (351) v 2654 (757) μm3, mean difference −51; 95% CI −687 to 585, p = 0.87).
Conclusions: These findings indicate hyperplasia of airway smooth muscle cells without hypertrophy and suggest that accumulation of airway smooth muscle cells may contribute to airway narrowing and bronchial hyperresponsiveness in CF.
- cystic fibrosis
- bronchial hyperresponsiveness
- airway smooth muscle
Statistics from Altmetric.com
Patients with cystic fibrosis (CF) manifest pulmonary infection and inflammation, bronchiectasis, and chronic airway obstruction. In some patients airway obstruction is reversible,1 and 40–50% have bronchial hyperresponsiveness.1,2 These lung function abnormalities overlap with those seen in asthma and suggest that smooth muscle remodelling may contribute to lung pathology in CF. Pathological studies in severe CF have demonstrated increased airway smooth muscle content.3,4 Whether increased smooth muscle content in CF is restricted to subjects with severe disease is unknown. Furthermore, whether these changes represent smooth muscle hyperplasia or hypertrophy is unknown. Differentiating hyperplasia from hypertrophy is germane to studies of the pathophysiology of smooth muscle changes in CF, because hyperplasia implies increased cell proliferation whereas hypertrophy implies an increase in the structural and contractile apparatus of individual cells. The goals of the current study were to apply bronchoscopy and design-based stereology to determine (1) whether an increase in the airway smooth muscle content is a feature of mild to moderate CF and (2) whether the increase is due to hyperplasia or hypertrophy.
METHODS
Subjects
We studied seven subjects with CF and 15 healthy control subjects. All CF subjects met Cystic Fibrosis Foundation criteria for the diagnosis of CF, had compatible lung disease, and sweat chloride testing results ranging from 82 to 120 mmol/l. Genotyping was performed for all subjects. Five subjects were homozygous for ΔF508, one subject was ΔF508/3905insT, and one subject was 1717-1/unidentified. Control subjects were healthy volunteers aged 18–55 with PC20 methacholine >16 mg/ml and forced expiratory volume in 1 second (FEV1) of >80% predicted. Subjects were excluded if they had a CF exacerbation or respiratory infection within the previous 6 weeks, smoking history (>10 pack years lifetime or any cigarette smoking in the last year), a history of haemoptysis requiring admission to an intensive care unit, or any other medical complication of CF that significantly increased the risk of sedation and bronchoscopy. Written informed consent was obtained from all subjects and the study was approved by the UCSF Committee on Human Research.
Data collection
Healthy control subjects completed two visits and patients with CF completed three visits. At visit 1 the procedures included medical history, physical examination, symptom questionnaire, spirometry, methacholine challenge testing, and allergen skin testing with a panel of 12 allergens as previously described.5 At visit 2 bronchoscopy was performed and bronchial biopsy specimens taken as previously described.6 Subjects with CF came for an additional safety visit 1 week after bronchoscopy for spirometric testing and a physical examination. Spirometric tests were performed according to ATS criteria using a dry rolling seal spirometer (Model VRS2000, PDS Instrumentation, S & M Instrument Co, Doylestown, PA, USA).
Bronchoscopy
During bronchoscopy up to six bronchial biopsy specimens were obtained for morphometric analysis from second to fourth order carina using a spiked fenestrated forceps (Pentax KH2411S and Pentax 8228, Pentax Precision Instrument Corporation, Orangeburg, NY, USA) yielding biopsies of 1–2 mm in diameter.
Tissue morphometry
Morphometry was performed using methods from design-based stereology.7 The biopsy specimens were formalin fixed and embedded in paraffin in a spherical mould to introduce isotropic uniform random (IUR) orientation as described previously.7,8 The smooth muscle volume fraction was measured using point and line intersection counting at 870× total magnification in sections stained for α-smooth muscle actin (Clone 1A4, NeoMarkers, Fremont, CA, USA) and counterstained with Gill’s #3 haematoxylin (Fisher Scientific, Hampton, NH, USA) as previously described.6 Briefly, the number of points overlying smooth muscle and other submucosal tissue and the number of lines intersecting the apical surface of the basal lamina (zona reticularis) by light microscopy were recorded by an investigator (PGW) who was blinded to the study. These counts allow calculation of the volume of smooth muscle per volume of submucosa and the volume of smooth muscle per surface area of basal lamina (the three dimensional analogue of perimeter length of the basement membrane3) using equations from design-based stereology.7 Smooth muscle cell number and size were measured using the physical dissector method at 700× total magnification applied to serial sections stained with Gill’s haematoxylin #3 (Fisher Scientific) and eosin Y (Sigma Diagnostics, St Louis, USA) as previously described.6 Systematic random sampling across the entire cross section of all of the biopsy specimens obtained from each subject was facilitated by a stereology microscopy system (CAST-Grid System, Olympus, Denmark). Volume fraction measurements were made on a mean of 67 fields and a mean of 240 smooth muscle point counts (range 95–328) per subject. The smooth muscle cell number was estimated using a mean of 38 fields per subject (range 20–46). One subject yielded insufficient smooth muscle for cell size estimation. Measurements of airway smooth muscle in healthy subjects were previously reported in a study of smooth muscle remodelling in asthma.6
Statistical analysis
Smooth muscle volume fraction and mean cell size in subjects with CF and in controls were compared using an unpaired t test, and measurements of cell number were compared using the Wilcoxon rank-sum test. Relationships between morphometric measures and lung function or allergen skin prick testing results were assessed using Pearson’s correlation. Data are presented as mean (SD) or median (interquartile range, IQR). Data analysis was performed with STATA 5.0 (StataCorp, College Station, TX, USA) using two sided tests; a p value of <0.05 was considered statistically significant.
RESULTS
Subjects with CF were similar to control subjects in age (mean 28 v 31 years, mean difference 3.5; 95% CI −2.4 to 9.3, p = 0.22), sex (5F:2M v 7F:8M, p = 0.38) and number of positive allergen skin prick tests (2 (IQR 0–6) v 2 (IQR 0–6), p = 0.64). Subjects with CF had reduced lung function with a mean pre-bronchodilator forced expiratory volume in 1 second (FEV1) of 60% predicted (range 45–71%) and forced vital capacity (FVC) of 81% predicted (range 66–100%). There was no significant difference in improvement in FEV1 after bronchodilator between groups (% change 4.0% in CF subjects and 2.8% in controls, mean difference −1.2; 95% CI −5.4 to 2.9, p = 0.55). PC20 methacholine was performed for only three subjects with CF because of safety concerns based on pre-challenge lung function testing (median PC20 = 64 (range 20–64) for healthy subjects v 1.12 (0.47–11.52) for subjects with CF).
The surface area of basal lamina per volume of submucosa was similar in CF subjects and normal control subjects (0.0105 (0.0030) v 0.0100 (0.0023) μm2/μm3, mean difference −0.0005; 95% CI −0.0029 to 0.0020, p = 0.69). The volume of smooth muscle in the airway submucosa of CF subjects was 1.63 times higher than normal when indexed to the volume of submucosa (0.173 (0.08) v 0.106 (0.042) mm3/mm3, mean difference −0.067; 95% CI −0.12 to −0.013, p = 0.017, fig 1) and 1.79 times higher than normal when indexed to the surface area of basal lamina (20.0 (6.7) v 11.2 (5.1) mm3/mm2, mean difference −8.8; 95% CI −14.1 to −3.4, p = 0.003). However, the mean volume of individual smooth muscle cells was nearly identical in CF subjects and healthy controls (fig 1). Using these values, the calculated number of smooth muscle cells per surface area of basal lamina was numerically greater in subjects with CF but the differences were not statistically significant (median 6618 (IQR 3842–7741) v 4236 (IQR 2076–6236) cells/mm2, p = 0.18). In the seven subjects with CF there was no significant relationship between any measure of smooth muscle volume or number and lung function, expressed as % predicted FEV1 or FEV1/FVC (all p>0.1 by Pearson’s correlation). Interestingly, the volume of smooth muscle per volume of submucosa correlated with the number of positive allergens on skin prick testing in CF subjects (r = 0.81, p = 0.027, Pearson’s correlation).
{kind=link}
Volume fraction of smooth muscle in the airway submucosa (left panel) and mean volume of individual smooth muscle cells (right panel) in bronchial biopsy specimens from subjects with cystic fibrosis (CF) compared with healthy control subjects.
DISCUSSION
Previous pathological studies have demonstrated increased airway smooth muscle content in patients with severe CF.3,4 The current study extends these findings by showing that the smooth muscle content of the airway in subjects with CF and mild to moderate airway obstruction is increased compared with healthy controls, and that this increase is due to smooth muscle cell hyperplasia without hypertrophy. These findings imply that smooth muscle cell proliferation is a characteristic of airway remodelling in CF. Although relationships between structural changes to airway smooth muscle and abnormalities of lung function were not evident in our small clinical sample of subjects with CF (n = 7), increased smooth muscle content in the airway in CF could contribute to airway hyperresponsiveness by increasing the force of contraction in response to agonist, as suggested by a previously published mathematical model.3
Airway smooth muscle remodelling occurs in asthma9 and, based on similar mathematical models, may contribute to airway narrowing and hyperresponsiveness.10 Using the same design-based stereological approach applied here, we recently found that mild to moderate asthma is also characterised by smooth muscle cell hyperplasia without hypertrophy.6 The similarities between smooth muscle cell changes in asthma and in CF suggest parallels between these two diseases in mechanisms of remodelling and airway hyperresponsiveness.
The smooth muscle changes in CF may reflect the direct effects of ongoing airway inflammation. The inflammatory cell infiltrate in CF is predominantly neutrophilic11 and reactive oxygen species—potential products of neutrophils—have recently been proposed to stimulate smooth muscle cell proliferation.12 Alternatively, it is possible that inflammatory cells and mediators commonly associated with the allergic inflammation of asthma contribute to the inflammatory milieu and structural changes in the airway in CF. Indeed, previous studies of reversible airway obstruction and bronchial hyperesponsiveness in CF have found a relationship between these lung function abnormalities and atopy or elevated IgE,2,13 although other studies have not confirmed these findings.1,14 In our small sample we found a positive correlation between smooth muscle content and number of positive allergen skin prick tests in subjects with CF.
Alternatively, the smooth muscle changes in CF may represent a response to airway epithelial cell injury and repair processes. Epithelial alterations that have been noted in pathological studies of CF include loss of cilia and stratification, and squamous metaplasia.15 The possibility that local mediators released during dysfunctional epithelial repair processes lead to the pathological changes in the submucosa in asthma has been articulated as the theory of “epithelial-mesenchymal trophic unit”.16 It is possible that a similar mechanism explains our observed changes in smooth muscle in subjects with CF.
The use of bronchoscopically obtained biopsy specimens has some limitations. We sampled only relatively large airways (second to fourth order bronchi) and could not measure changes in the distal airway. In addition, we only took biopsy specimens at the carinae so we cannot fully account for morphological changes that may be focally distributed elsewhere.
In conclusion, we have found that subjects with CF and mild to moderate airway obstruction have increased smooth muscle content in the airway with normal smooth muscle cell size. These findings suggest that factors contributing to the accumulation of airway smooth muscle cells, such as smooth muscle cell proliferation, contribute to the pathological changes in the airway in CF and may contribute to chronic airway narrowing or airway hyperresponsiveness.
REFERENCES
Footnotes
-
This study was supported by a pilot study grant from the Cystic Fibrosis Research and Development Program, a Cystic Fibrosis Foundation Fellowship Award (SRH), by NIH grants K23 RR17002, and RO1 HL61662 and by the General Clinical Research Center, Moffitt Hospital, UCSF, with funds provided by the NCRR, 5 MO1 RR-00079, US Public Health Service.
Linked Articles
- airwaves