Article Text

Abstract
Background The inhaled corticosteroid fluticasone furoate (FF) in combination with the long-acting β2 agonist vilanterol (VI) is in development for asthma and chronic obstructive pulmonary disease.
Objective To assess the safety and tolerability of FF/VI over 52 weeks in patients with asthma.
Methods Patients (aged ≥12 years; on inhaled corticosteroid) were randomised (2:2:1) to FF/VI 100/25 µg or FF/VI 200/25 µg once daily in the evening, or fluticasone propionate (FP) 500 µg twice daily. Safety evaluations included adverse events (AEs), non-fasting glucose, potassium, 24-h urinary cortisol excretion, ophthalmic assessments, heart rate and pulse rate.
Results On-treatment AEs were similar across groups (FF/VI 66–69%; 73% FP). Oral candidiasis/oropharyngeal candidiasis was more common with FF/VI (6–7%) than FP (3%). Twelve serious AEs were reported; one (worsening hepatitis B on FP) was considered drug related. Statistically significant cortisol suppression was seen with FP compared with both FF/VI groups at Weeks 12 and 28 (ratios [95% CI] to FP ranged from 1.43 [1.11 to 1.84] to 1.67 [1.34 to 2.08]; p≤0.006), but not at Week 52 (ratios to FP were 1.05 [0.83 to 1.33] for FF/VI 100/25 µg and 1.09 [0.87 to 1.38] for FF/VI 200/25 µg). No clinically important changes in non-fasting glucose, potassium, QT interval corrected using Fridericia's formula (QTc[F]) or ophthalmic assessments were reported. Pulse rate (10 min post dose [Tmax], Week 52) was significantly increased with FF/VI versus FP (3.4 bpm, 95% CI 1.3 to 5.6; p=0.002 [FF/VI 100/25 µg]; 3.4 bpm, 95% CI 1.2 to 5.6; p=0.003 [FF/VI 200/25 µg]). Mean heart rate (24-h Holter monitoring) decreased from screening values in all groups (0.2–1.1 bpm FF/VI vs 5 bpm FP; Week 52).
Conclusions FF/VI (100/25 µg or 200/25 µg) administered once daily over 52 weeks was well tolerated by patients aged ≥12 years with asthma. The overall safety profile of FF/VI did not reveal any findings of significant clinical concern.
ClinicalTrials.gov NCT01018186
- Asthma
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Key messages
What is the key question?
-
Does the once-daily inhaled corticosteroid/long-acting β2 agonist combination fluticasone furoate/vilanterol (FF/VI) have an acceptable safety and tolerability profile over 52 weeks in patients (≥12 years) with asthma?
What is the bottom line?
-
Over 52 weeks, FF/VI (100/25 µg and 200/25 µg) was well tolerated with an acceptable safety profile and there were no findings of significant clinical concern.
Why read on?
-
The doses of once-daily FF/VI selected in this study (100/25 µg, 200/25 µg) were based on efficacy and safety/tolerability data from short-term (≤8 weeks) phase II dose-ranging studies of FF and VI in asthma. Results from this phase III study provide information on the longer-term effects of FF/VI on a number of clinically meaningful safety and tolerability endpoints such as UC excretion, ocular changes and heart rate.
Introduction
Inhaled corticosteroids (ICS) are recommended for patients with symptomatic asthma taking as-needed short acting bronchodilators.1 ,2 Long-acting β2 agonists (LABA) are recommended in combination with ICS for patients who remain symptomatic on low–medium dose ICS alone.1–3 The ICS, fluticasone furoate (FF), together with the LABA, vilanterol (VI), are in development as a novel, single-inhaler, once-daily ICS/LABA combination for asthma and chronic obstructive pulmonary disease. By simplifying the dosing regimen, FF/VI could potentially increase patient adherence and thereby control of asthma symptoms.4 Results from three dose-ranging phase II studies with FF once daily in asthma (25–200 µg;5 100–400 µg;6 200–800 µg7) show that FF doses of 100 µg and 200 µg are effective with an acceptable tolerability and safety profile across a range of asthma severities. Separate phase II studies with VI in asthma support administration of 25 µg once daily.8 ,9
The objectives of this study were to assess the safety and tolerability of 52 weeks of treatment with two strengths of inhaled FF/VI (100/25 µg and 200/25 µg) administered once daily in the evening in patients ≥12 years of age with asthma.
Patients and methods
Patients
Patients aged ≥12 years were eligible if they had asthma as defined by the National Institutes of Health criteria2 and were using ICS (500–1000 µg/day or equivalent) with or without additional controller medication for ≥4 weeks prior to Visit 1 (screening). Eligible patients had a best forced expiratory volume in 1 s of ≥50% of predicted normal at screening and could demonstrate forced expiratory volume in 1 s reversibility of ≥12% and ≥200 ml after salbutamol/albuterol inhalation or could provide documented history of the same reversibility reported within 12 months prior to screening. Patients had to stop their current ICS plus additional controller medication, where applicable, from the morning of Visit 2 (baseline). Patients were excluded if they had an exacerbation requiring corticosteroid treatment within 3 months of screening visit. Other exclusion criteria and prohibited medications are shown in the online supplementary material.
All patients gave written informed consent. The study was approved by local ethics review committees and was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines and all applicable regulatory requirements.
Study design and treatments
This phase III, randomised, multicentre, double-blind, double-dummy, active comparator, parallel group study (HZA106839; ClinicalTrials.gov identifier NCT01018186) was conducted in 45 centres in four countries (USA, Germany, Ukraine, Thailand), between 19 October 2009 and 12 May 2011.
Patients meeting the entry criteria entered a 2-week run-in period for completion of baseline safety evaluations and asthma status measures. After run-in, patients were randomised (2:2:1) to one of three treatments: FF/VI 100/25 µg (representing an emitted dose of 92/22 µg) or FF/VI 200/25 µg (emitted dose 184/22 µg) once daily in the evening or fluticasone propionate (FP) 500 µg twice daily for 52 weeks. FF/VI was administered via a dry powder inhaler (DPI) and FP was administered by DISKUS/ACCUHALER. To maintain blinding of the treatment schedule, a double-dummy system was employed with a placebo DISKUS/ACCUHALER administered to patients receiving FF/VI and a placebo DPI administered to patients receiving FP. Following randomisation, patients attended clinic during Weeks 2, 4, 8, 12, 20, 28, 36, 44 and 52. A follow-up assessment was performed 1 week after completing study medication. Adherence with study treatment (using the device dose counters) was assessed throughout the treatment period.
The central randomisation schedule was generated by the sponsor using a validated computerised system (RandALL; GlaxoSmithKline, UK). Patients were randomised using an automated, telephone-based registration and medication-ordering system (Registration and Medication Ordering System [RAMOS]).
Endpoints
The incidence of adverse events (AEs), serious AEs (SAEs) and severe asthma exacerbations were recorded throughout the study. A severe asthma exacerbation was defined according to American Thoracic Society/European Respiratory Society taskforce guidelines as deterioration of asthma requiring the use of systemic corticosteroids for ≥3 days, or an inpatient hospitalisation or emergency room visit due to asthma that required systemic corticosteroids.10 The rate of oral/oropharyngeal candidiasis was diagnosed by oral examination (each visit); where the investigator suspected oral/oropharyngeal candidiasis a confirmatory culture specimen was taken. A positive culture was required for oral/oropharyngeal candidiasis to be reported as an AE.
Clinical laboratory assessments, including non-fasting glucose, potassium, liver function tests and 24-h urinary cortisol (UC) excretion were completed prior to treatment and at Weeks 12, 28 and 52 or early withdrawal. UC was measured by an inhouse liquid chromatography tandem mass spectrometry method (Quest Diagnostics, Valencia, California, USA). Electrocardiogram (ECG) assessment (heart rate [HR] and QTc[F], each measured 10 min post dose) was completed at screening, at Weeks 2, 12, 28 and 52 or early withdrawal. Holter monitoring (subset of ∼50% patients) for 24 h was completed at screening, baseline, and Weeks 28 and 52 or early withdrawal. Vital signs (systolic and diastolic blood pressure, pulse rate) were measured prior to treatment and 10 min post dose at every visit.
Ophthalmic assessments were completed by a qualified ophthalmologist at screening, Week 28 and Week 52 or early withdrawal. Examinations of posterior subcapsular opacity, cortical opacity, nuclear colour and nuclear opalescence were conducted using the Lens Opacities Classification System, Version III (LOCS III).11 Separate assessments for intraocular pressure and visual acuity (LogMAR) were also undertaken.
Plasma concentrations of FF and VI and derived pharmacokinetic parameters were reported, however, they are not presented in this manuscript.
Statistical analysis
With safety the primary objective of the study, formal sample size calculation was not performed. The recruitment target was determined based on International Conference Harmonisation guidelines and feasibility considerations. Randomisation was planned for 200 patients to each FF/VI group, and 100 patients to the FP group. Assuming a maximum 30% withdrawal rate during the 52-week treatment period, it was expected at least 140 patients in each FF/VI group and 70 patients in the FP group would have exposure data for the full 52 weeks. Further information regarding the sample size estimation is provided in the online supplementary material.
The intent-to-treat (ITT) population comprised all patients randomised to treatment who received at least one dose of study drug (primary population for all data displays). The UC population consisted of patients in the ITT population whose urine samples at baseline and at least one post-baseline assessment did not have confounding factors that would affect interpretation of results (see online supplementary material). Statistical analyses were performed for 24-h UC, HR, QTc(F), blood pressure and pulse rate. The relative safety of each FF/VI dose was evaluated by assessing the confidence intervals for the treatment differences compared with the marketed benchmark, FP 500 µg twice daily for these endpoints and p values were provided for descriptive purposes. For all other endpoints, summary statistics were evaluated. Detailed descriptions of all analyses and statistical tests used are provided in the online supplementary material.
Results
Study population
A total of 503 patients were randomised (ITT population); 393 (78%) completed the study (figure 1). Most withdrawals took place between Weeks 12 and 36 (Days 84–252) (figure 2). Treatment groups were well matched with respect to baseline demographics, exacerbation history (previous 12 months) and screening lung function (table 1). Mean age overall (ITT population) was 39 years; 16% of patients were adolescents (aged 12–<18 years).
Patient baseline demographics and screening lung function (intent-to-treat population)

CONSORT/patient flow diagram.

Patient withdrawal from the study over time (intent-to-treat population).
Mean treatment compliance with DPI and DISKUS was >96%. Further information on compliance and inter-visit compliance are provided in the online supplementary material.
Adverse events
The incidence of on-treatment AEs was similar across treatment groups (66–73%), with headache, upper respiratory tract infection and nasopharyngitis the most frequently reported (table 2). Treatment-related AEs (as defined by investigator) were reported by 13−14% of patients across treatment groups, with oral/oropharyngeal candidiasis, dysphonia and extrasystoles the most frequently reported (table 2). Lower respiratory tract infection, pneumonia and ‘any ocular effect’ (predefined AEs of special interest) occurred with low and similar frequency across the groups. Cardiovascular effects were more common on FF/VI 200/25 µg (18%) versus FF/VI 100/25 µg (12%) and FP (10%) due to extrasystoles reported on FF/VI 200/25 µg. Twelve patients reported a SAE; 11 of these were on treatment, three (1%) receiving FF/VI 100/25 µg, one (<%1) receiving FF/VI 200/25 µg and seven (7%) receiving FP (table 2), and one was post-treatment (spontaneous abortion) on FF/VI 100/25 µg. One patient had a SAE in the FP group (worsening hepatitis B) that was considered by the investigator to be possibly treatment related. Five of the 11 patients reporting an on-treatment SAE were withdrawn from the study due to the SAE. No deaths were reported during the study.
Overview of AEs and the most common (≥3%) on-treatment AEs and the most common (≥2%) treatment-related AEs (intent-to-treat population)
Exacerbations
Three patients (1%) on FF/VI 100/25 µg, six (3%) on FF/VI 200/25 µg and three (3%) on FP experienced a severe exacerbation during the treatment period. Three patients (one on FF/VI 100/25 µg and two on FP) were hospitalised as a result (none required intubation).
24-h urinary cortisol
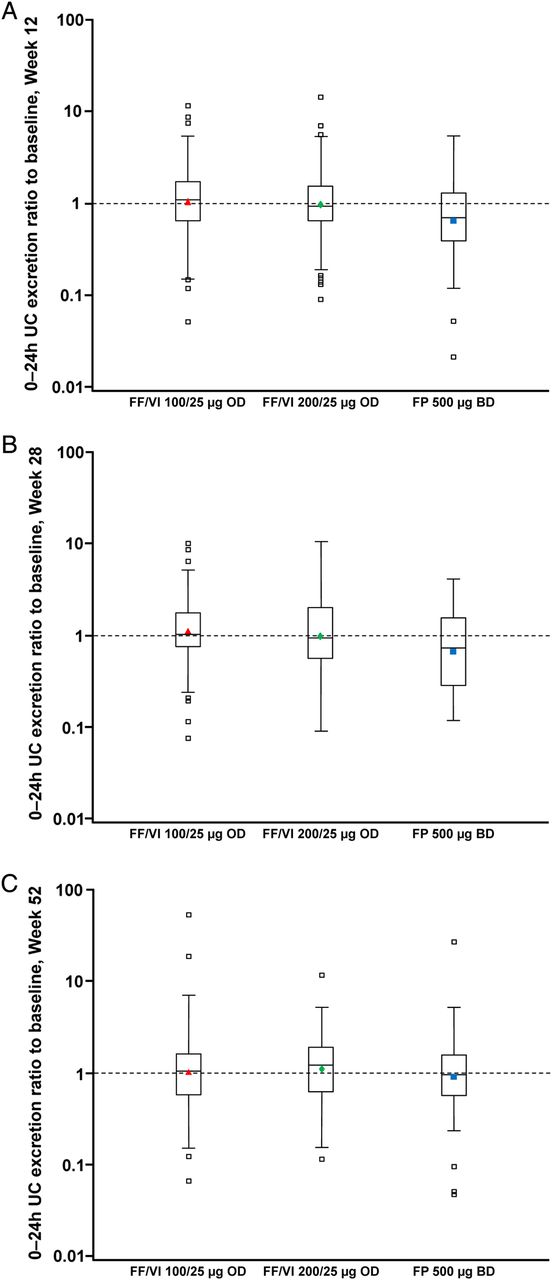
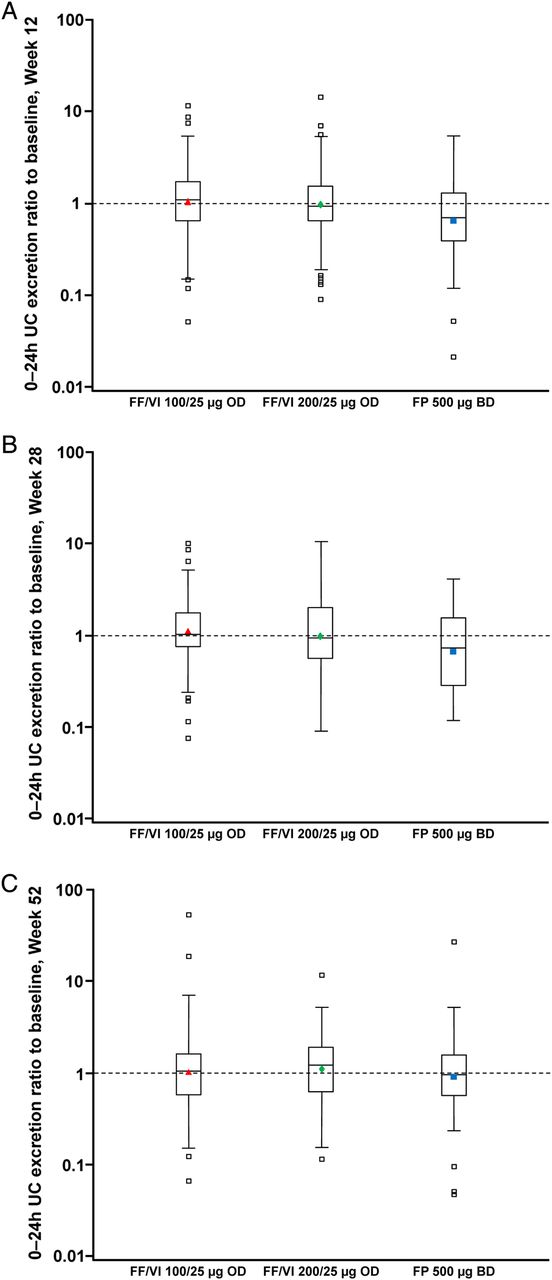
UC measurements were available in 362 patients. The 24-h UC excretion (nmol/24 h) ratios to baseline with FF/VI indicated little change from baseline values at Weeks 12, 28 and 52; for FP, the ratios indicated a reduction in UC excretion from baseline values at Weeks 12 and 28 but not at Week 52 (figure 3). Statistically significant (p≤0.006) cortisol suppression was seen with FP compared with FF/VI 100/25 µg and FF/VI 200/25 µg, respectively, at Week 12 (ratio to FP: 1.67 [95% CI 1.34 to 2.08]; 1.52 [1.22 to 1.89]) and Week 28 (1.65 [1.29 to 2.13]; 1.43 [1.11 to 1.84]), but not at Week 52 (1.05 [0.83 to 1.33]; 1.09 [0.87 to 1.38]). Using information from the ITT population, the majority of patients had ‘normal’ or ‘no change’ from baseline in 24-h urine-free cortisol excretion at any post-baseline visit (76–81% FF/VI; 70% FP; table 3; see online supplementary figure S1).
Urinary cortisol excretion outside the normal range* and change from baseline relative to the normal range (intent-to-treat population)
{kind=link}
{kind=link}
{kind=link}
24-h urinary cortisol (UC) excretion ratio to baseline at (A) Week 12, (B) Week 28 and (C) Week 52 (UC population).
Laboratory assessments
Mean values for clinical chemistry parameters were similar across the treatment groups at screening and at Weeks 12, 28 and 52. No important safety concerns were reported for liver chemistry analyses. No clinically important increases in non-fasting glucose or decreases in potassium were reported. The mean (standard deviation) change from baseline in glucose (mmol/l) at Week 52 was 0.45 (1.839) with FF/VI 100/25 µg, 0.11 (1.426) with FF/VI 200/25 µg and −0.20 (1.191) with FP. The mean change from baseline in potassium (mmol/l) at Week 52 was −0.18 (0.461) with FF/VI 100/25 µg, −0.12 (0.535) with FF/VI 200/25 µg and −0.10 (0.446) with FP.
Vital signs
No clinically significant safety concerns were reported in systolic or diastolic blood pressure readings. Patients in the FF/VI groups had increases from baseline in mean pulse rate over clinic visits, whereas patients in the FP group generally had decreases from baseline in pulse rate. Statistical analysis of changes from baseline in pulse rate at Week 52 showed significant increases in pulse rate (10 min post dose; [Tmax]) with FF/VI 100/25 µg (3.4 bpm; 95% CI 1.3 to 5.6; p=0.002) and with 200/25 µg (3.4 bpm; 95% CI 1.2 to 5.6; p=0.003) versus FP. However, examination of patients with increases in pulse >20 bpm revealed considerable inter-visit variability; increases occurred sporadically (usually at one or two visits) during the study and no treatment-related patterns were evident.
ECG
Statistically significant increases in HR with FF/VI were also reported versus FP (differences between FF/VI and FP ranged from 4.1 bpm [95% CI 1.9 to 6.4] to 5.9 bpm [95% CI 3.3 to 8.5]; p<0.001). Differences between FF/VI and FP in adjusted mean change from baseline in QTc(F) were –1.4 to 1.2 msec and none were statistically significant. Maximum post-baseline values and change from baseline in QTc(F) are summarised in table 4.
Maximum post-baseline values and change from baseline in QTc(F), and mean (0–24 h) HR by Holter monitor by visit for patients with ≥16 h of recorded data (intent-to-treat population)
24-h Holter monitoring
Mean HR at post-baseline visits were similar to screening and no increases were reported; all groups had a slight decrease in HR with the greatest reduction reported on FP (5 bpm at Weeks 28 and 52 vs screening value; table 4). Approximately 50% of patients in each treatment group had no reported ventricular ectopics at screening (table 5). The median number of ventricular ectopics ranged from 0–1 across the treatment groups. The proportion of patients with >50 ventricular ectopics (any type) and >1 ventricular singlet was higher in the FF/VI groups compared with the FP group. The incidence of ≥1 ventricular couplet was low and generally similar across treatment groups. Non-sustained ventricular tachycardia (NSVT; >100 bpm, 3−30 beats) was observed in two patients (2%) from each FF/VI group but not with FP. Sustained supraventricular tachycardia (>100 bpm, >30 beats) was only observed in the FF/VI 200/25 µg group (three patients [3%]). All events of NSVT and sustained supraventricular tachycardia resolved without treatment. Further information on the observed non-sustained and sustained tachycardia is reported in the online supplementary material.
Ventricular ectopics by visit for patients with ≥16 h of recorded data (intent-to-treat population)
Ophthalmic assessments
No clinically significant differences between groups on the ophthalmic assessments of subcapsular opacity, cortical opacity, nuclear colour, nuclear opalescence, intraocular pressure or visual acuity were reported (see online supplementary material for further details).
Discussion
The results from this 52-week study of FF/VI in adolescents and adults with asthma showed that FF/VI 100/25 µg or 200/25 µg, dosed once daily in the evening, was well tolerated. We compared the safety and tolerability profile following FF/VI to that obtained with FP 500 µg twice daily. FP 500 µg has been shown to improve lung function in patients with asthma and 500 µg twice daily is the highest recommended dosage for treating patients with asthma symptomatic on low-dose ICS.12 In an earlier phase II study, FP 500 µg twice daily had a similar efficacy to FF 200 µg once daily (vs placebo) in patients with persistent moderate-to-severe asthma.7 However, FP 500 µg twice daily is associated with more steroid-associated AEs than lower FP doses13–15 and this dose of FP was therefore selected in our study as a benchmark to which FF/VI safety data could be compared.
The patient population contained a wide age range (12–73 years) in order to adequately assess the safety and tolerability of FF/VI in adolescent and adult patients with asthma. In addition, the majority of patients had not experienced an asthma exacerbation (all severities) in the past 12 months.
The number of withdrawals due to an AE was similar across treatment groups (figure 1). The most frequently reported AE was headache and upper respiratory tract infection, and the frequency was generally similar across treatment groups. The frequency of SAEs was also similar across treatment groups. One SAE with FP (worsening hepatitis B) was considered by the investigator to be possibly treatment related. However, this SAE on FP is not substantiated by reports in the current literature and has not been identified as a potential AE associated with FP therapy in asthma.12
More patients on FF/VI reported the AE of oral/oropharyngeal candidiasis than those receiving FP. However, the lower rate of oral/oropharyngeal candidiasis with FP versus FF/VI is not consistent with rates of this AE in other studies of FP when dosed at 500 µg twice daily.13 ,14 Additionally, in earlier phase II studies of FF in asthma, the incidence of oral candidiasis was generally similar to FP at doses of FF ≤600 µg once daily.5–7 Other potential effects of long-term ICS use include increased intraocular pressure and cataract formation. Extensive study-specific ophthalmic examinations reported no apparent effects with FF/VI or FP.
There was little change from baseline in UC excretion in any treatment group. However, UC ratios to baseline in patients on FP were lower compared with FF/VI (figure 3). The reduction from baseline UC with FP was significantly different compared with FF/VI at Weeks 12 and 28 but not at Week 52 suggesting that UC excretion had recovered to pre-treatment levels in these patients (UC population). Prior to randomisation the majority of patients were receiving medium-dose ICS (data not shown), therefore FP 500 µg twice daily (high dose) may have been a ‘step-up’ for these patients. Although not formally evaluated in this study, such a ‘step-up’ in treatment may account, at least in part, for the observed excretion ratios to baseline with FP observed at Weeks 12 and 28 in this study. However, it is important to note that the majority of patients had ‘normal’ or ‘no change’ in 24-h UC excretion at any post-baseline visit. Treatment compliance was very high throughout the study (including inter-visit assessments), suggesting that changes in patients’ level of compliance to FP study medication during the study is an unlikely reason for the UC excretion observations reported with FP at Weeks 12, 28 and 52.
Statistically significant differences with FF/VI versus FP were reported for HR (by ECG) and pulse rate (both measured at Tmax). However, the increases in pulse rate were reported only sporadically during the study (one or two visits only). Furthermore, the ECG HR findings observed post dose are consistent with that observed with other inhaled agonists at the β-agonist receptor. No increases in HR by 24-h Holter monitoring were observed, and the increases in HR by ECG, although statistically significant versus FP, were not considered to be clinically significant. No clinically significant effects on QTc(F) outputs were observed during the study with FF/VI versus FP. Indeed, adjusted mean change from baseline in QTc(F) with FF/VI 100/25 µg ranged from 1.2 to 5.7 FF/VI 200/25 µg and for FF 200/25 µg was –0.8 to 3.8 msec. The QTc(F) results for FF/VI 200/25 µg are similar to results from a placebo-controlled cardiac safety study (FF/VI 800/100 µg and 200/25 µg) in healthy subjects where time-matched mean differences from placebo with FF/VI 200/25 µg in QTc(F) were <5 msec.16 NSVT was reported in 2% of patients on FF/VI 100/25 µg and FF/VI 200/25 µg and no patients on FP. However, a 2% incidence is not greater than the expected (‘background’) rate of 1–3% in healthy subjects.17–19 These episodes of NSVT were isolated and short (3–7 beats) with only one patient having two episodes (3 and 4 beats). No clinically relevant decreases in potassium or increases in non-fasting glucose were observed.
An important strength of this study is the 52-week duration which provides useful information on the longer-term safety and tolerability effects of FF/VI in patients with asthma. Another is the double-dummy, double-blind design, which avoids bias in reporting of AEs. Measuring HR 10 min post dose following FF/VI administration is an advantage as this permitted evaluation of the maximum potential effect of VI (at the Tmax) on HR. A study limitation is that more information on treatment effects on pulse rate and HR (by ECG) would have been collected had measurements been taken over a 24-h period post dose, not just at 10 min post dose. Another is that we did not include an active LABA comparator; however current treatment guidelines1 recommend against the use of LABA monotherapy in asthma due to a possible increased risk of severe exacerbation. Also, some patients in the study had moderate asthma so for these patients the FP dose was very high (1000 µg/day). However, a lower daily dose of FP for 52 weeks would have been a ‘step down’ for the majority of patients who had more severe asthma.
Conclusion
In this study, both doses of FF/VI (100/25 µg and 200/25 µg) administered once daily over 52 weeks were well tolerated by patients ≥12 years of age with asthma. The overall safety profile observed with FF/VI did not reveal unusual or unexpected findings of significant clinical concern. Safety findings that were observed reflected the expected pharmacological activity of the ICS and LABA components.
Acknowledgments
This study was funded by GlaxoSmithKline. We thank all patients who took part in the study and all of the investigators at the 45 centres. Editorial support in the form of development of a draft outline in consultation with the authors, development of a manuscript first draft in consultation with the authors, editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments, copyediting, fact checking, referencing and graphic services was provided by Lisa Moore at Gardiner-Caldwell Communications and was funded by GlaxoSmithKline.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors All authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors, had full access to and interpreted the data, and wrote the manuscript. L Andersen, WH, JC and LJ developed the design and concept of the study. JC was the study statistician. All authors vouch for the accuracy and completeness of the data and the data analysis.
-
Funding The study was funded by GlaxoSmithKline. The fee for colour figures and open access was paid by GlaxoSmithKline.
-
Competing interests WWB has served as a consultant to AstraZeneca, Boehringer Ingelheim, Novartis and TEVA; served on advisory boards for Altair, Amgen, Centocor, GlaxoSmithKline, Johnson & Johnson, Merck Sharpe and Dohme and Pfizer; received lecture fees from Merck Sharpe and Dohme; and received research funding from AstraZeneca, Ception, GlaxoSmithKline, MedImmune and Novartis. PMO'B has served as a consultant to AstraZeneca, Almirall, Chiesi, and Novartis; has served on advisory boards for AIM, Altair, Boehringer Ingelheim, GlaxoSmithKline, Medimmune and Merck; has received lecture fees from Chiesi; and has received research funding from AstraZeneca, Asmacure, Altair, Amgen, Genentech, Topigen and Wyeth. ERB has served as a consultant to GlaxoSmithKline; and has performed clinical trials for GlaxoSmithKline, which have been administered by his employer Wake Forest University Health Sciences. JL has served as a consultant to and received lecture fees from AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme, Novartis and UCB Pharma; has been partly covered by some of these companies to attend previous scientific meetings including the ERS and the AAAAI; and has participated in clinical research studies sponsored by AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme and Novartis. AW has served as consultant to Almirall, AstraZeneca, Chiesi, Cytos, GlaxoSmithKline, Merck Sharpe and Dohme, Novartis; and has received lecture fees and research grants from Chiesi and GlaxoSmithKline. EDB has served as a consultant to and received lecture fees from GlaxoSmithKline; and his institution has received remuneration for participation in clinical trials sponsored by GlaxoSmithKline. WH was an employee of GlaxoSmithKline at the time the study was conducted and during the first drafts of the manuscript and now works for a Contract Research Organisation that undertakes work for pharmaceutical companies including GlaxoSmithKline. L Andersen, L Apoux, JC and LJ are employees of and hold stock in GlaxoSmithKline.
-
Ethics approval Local ethics review committees from the 45 centres involved in the study.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/