Article Text

Abstract
Background Cigarette smoking contributes to lung remodelling in chronic obstructive pulmonary disease (COPD). As part of remodelling, peribronchiolar fibrosis is observed in the small airways of patients with COPD and contributes to airway obstruction. Epithelial to mesenchymal transition (EMT) appears to be involved in the formation of peribronchiolar fibrosis. This study examines the EMT process in human bronchial epithelial cells (HBECs) from non-smokers, smokers and patients with COPD as well as the in vitro effect of cigarette smoke extract (CSE) on EMT.
Methods HBECs from non-smokers (n=5), smokers (n=12) and patients with COPD (n=15) were collected to measure the mesenchymal markers α-smooth muscle actin, vimentin and collagen type I and the epithelial markers E-cadherin, ZO-1 and cytokeratin 5 and 18 by real time-PCR and protein array. In vitro differentiated bronchial epithelial cells were stimulated with CSE.
Results Mesenchymal markers were upregulated in HBECs of smokers and patients with COPD compared with non-smokers. In contrast, epithelial cell markers were downregulated. In vitro differentiated HBECs underwent EMT after 72 h of CSE exposure through the activation of intracellular reactive oxygen species, the release and autocrine action of transforming growth factor β1, the phosphorylation of ERK1/2 and Smad3 and by the downregulation of cyclic monophosphate.
Conclusions The EMT process is present in bronchial epithelial cells of the small bronchi of smokers and patients with COPD and is activated by cigarette smoke in vitro.
- Airway Epithelium
- COPD Pathology
- Oxidative Stress
- Tobacco and the lung
Statistics from Altmetric.com
Key messages
-
What is the key question?
-
Are the bronchial epithelial cells from smokers and patients with COPD undergoing epithelial to mesenchymal transition (EMT)? Is cigarette smoke involved in bronchial EMT?
-
What is the bottom line?
-
Phenotypic markers of EMT are observed in bronchial epithelial cells of small bronchi from smokers and patients with COPD. Cigarette smoke induces EMT in differentiated bronchial epithelial cells via release and autocrine action of TGF-β1 as well as by enhancing oxidative stress. These results suggest that EMT could participate in the COPD remodelling process of small bronchi such as peribronchiolar fibrosis.
-
Why read on?
-
The EMT process is activated in the small bronchi of smokers and patients with COPD as part of the chronic lung remodelling process. Cigarette smoke directly increases mesenchymal transition in differentiated human bronchial epithelial cells.
Introduction
Chronic obstructive pulmonary disease (COPD) is characterised by airflow limitation that is progressive and not fully reversible. Cigarette smoking is considered the main risk factor for COPD and contributes to structural changes in the airways during COPD progression.1 Bronchial epithelial cells are the first anatomical barrier exposed to noxious particles of cigarette smoke and are involved in the initiation of airway remodelling through the release of several proinflammatory mediators, goblet cell hyperplasia and mucus hypersecretion.2 Other features that characterise COPD lung remodelling are the progressive loss of the alveolar wall, vascular remodelling with pulmonary hypertension and peribronchiolar fibrosis. As part of the fibrotic alterations in COPD, structural changes are observed primarily in the small airways (<2 mm in diameter), and the severity of the disease appears to be determined by thickening of the wall of the small airways caused by fibrosis and infiltration of inflammatory cells.3 However, the exact factor responsible for producing the remodelled matrix as well as the role of activated epithelium in the remodelling process and in fibroblast activation/differentiation into myofibroblasts in small airway fibrosis in COPD is unclear.
It has been established that myofibroblast accumulation may occur as a consequence of resident fibroblast to myofibroblast transition, airway smooth muscle to myofibroblast transition, epithelial to mesenchymal transition (EMT) or by the recruitment of circulating fibroblastic stem cells (fibrocytes).4 Depending on the precise physiological context, EMT can be induced either by environmental stresses/factors such as hypoxia5 and reactive oxygen species (ROS)6 or by extracellular mediators including transforming growth factor β1 (TGF-β1), fibroblast growth factor 2, connective tissue growth factor and insulin-like growth factor 2.7 Among these, TGF-β1, a multifunctional cytokine, has been shown to mediate EMT in vitro in several different cell lines including alveolar and bronchial epithelial cells and recently in pseudostratified bronchial epithelial cells.8 In vivo evidence for TGF-β1-mediated EMT has also been reported, confirming the master regulation role of TGF-β1.9
In epithelial cells, TGF-β1-induced EMT is mediated by several downstream factors such as ROS generation, activation of mitogen-activated protein kinase and Smad signalling. Thus, antioxidant treatments6 and other treatments directed at attenuating intracellular ROS, such as the increase in intracellular adenosine 3′,5′-cyclic monophosphate (cAMP),10 effectively inhibit TGF-β1-induced EMT.
It has recently been shown that cigarette smoke may induce EMT in the alveolar type II cell line A54911 and the bronchial epithelial cell line BEAS2B,12 but the effect of cigarette smoke on primary human bronchial epithelial cell (HBEC) to mesenchymal transition is currently poorly understood. In addition, evidence for EMT in the large airways of smokers and patients with COPD has been suggested by Sohal and colleagues, since cells included in the reticular basement membrane of large airways showed EMT markers.13 However, whether EMT is present in the small airways of smokers and patients with COPD and whether cigarette smoke directly induces EMT in differentiated HBECs remains to be elucidated.
Using primary HBECs from small human bronchi of non-smokers, smokers and patients with COPD, we studied typical mesenchymal and epithelial cell markers of the EMT process. Furthermore, we determined whether cigarette smoke may induce EMT in differentiated air–liquid interface cultures as well as the potential signalling mechanisms involved.
Methods
A more detailed version of the methods is given in the online supplement.
Patients
Human lung tissue from non-smokers (n=5), smokers (n=12) and patients with COPD (n=15) was obtained from patients undergoing surgery for lung carcinoma who gave informed consent, as previously described.14 Inclusion and exclusion criteria are defined in the online supplement.
Isolation of primary bronchial epithelial cells
Primary HBECs were isolated and cultured from small pieces of human bronchi (0.5–1 mm internal diameter) as previously outlined15 and cultured for 1 week to analyse mRNA and protein expression (see online supplement for details).
Immunohistochemistry and immunofluorescence
Immunohistochemical analysis of α-smooth muscle actin (α-SMA), collagen type I, vimentin, NOX4, E-cadherin and ZO-1 was performed in human pulmonary tissue using mouse anti-human α-SMA (catalogue no A5228; Sigma), rabbit anti-human collagen type I antibody (catalogue no PA1-26204; Affinity Bioreagents), mouse anti-human vimentin (catalogue no V6389; Sigma), NOX4 (catalogue no NB110-58849; Novus Biologicals), mouse anti-human E-cadherin (catalogue no CM1681; ECM BioScience) and rabbit anti-human ZO-1 (catalogue no ab59720; Abcam). The scores of the staining intensity (0–3) and the percentage of immunoreactive cells (1–4 where 1 represents 0–25% cells positive, 2 represents 26–50% positive, 3 represents 51–75% positive and 4 represents 76–100% positive) were then multiplied to obtain a composite score ranging from 0 to 12. Secondary antibody anti-rabbit/mouse rhodamine/FITC was used for immunofluorescence analysis of primary bronchial epithelial cells. To detect F-actin distribution, phalloidin-tetramethylrhodamine isothiocyanate was employed (see online supplement for full details).
Culture of air–liquid interface bronchial epithelial cells
Differentiated HBECs for in vitro experiments were obtained from non-smoker patients and cultured in air–liquid interface until pseudostratified bronchial epithelium was obtained.15 Details are described in the online supplement.
Preparation of cigarette smoke extract and transepithelial electrical resistance assay
Cigarette smoke extracts (CSE) were obtained as previously outlined16 using a research cigarette (2R4F; Tobacco Health Research, University of Kentucky, Kentucky, USA). Transepithelial electrical resistance (TEER) was assayed using a millicell ERS-2 epithelial volt-ohm meter. Details of the TEER assay and the method of obtaining CSE, in vitro stimulation conditions as well as drug incubations are described in the online supplement.
Real-time PCR
Real-time PCR was performed as previously described.17 Relative quantification of different transcripts was determined by the 2−ΔΔCt method using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an endogenous control and with normalisation to the non-smoker group or control group as appropriate (see online supplement for further details).
Protein array and western blot analysis
Protein expression in lung tissue samples from non-smokers, smokers and patients with COPD was analysed with Zeptosens (Division of Bayer (Schweiz), Switzerland) protein array technology as previously outlined.14 Western blot analysis was used to analyse β-catenin expression in the nucleus and cytoplasm (see online supplement for further details).
DCF fluorescence measurement of reactive oxygen species
2′,7′-Dichlorodihydrofluorescein diacetate (H2DCF-DA, Molecular Probes, Nottingham, UK) was used to monitor the intracellular ROS in differentiated HBECs as previously described14 (see online supplement for details).
Enzyme-linked immunosorbent assays for TGF-β1, cAMP and MMP-9
Analyses of active TGF-β1 and intracellular cAMP were performed using a Quantikine Human TGF-β1 Immunoassay (R&D Systems, Madrid, Spain) and cAMP Biotrak enzyme immunoassay system (Amersham, London, UK) according to the manufacturers’ instructions. Matrix metalloproteinase (MMP)-9 was determined using an assay kit for MMP-9 (R&D Systems, Nottingham, UK) according to the manufacturer's protocol (see online supplement for further details).
Statistics
Statistical analysis of results was carried out by parametric or non-parametric analysis as appropriate. Data from non-parametric analysis were expressed as median, IQR and minimum and maximum values. The Mann–Whitney U test or Kruskal–Wallis test followed by Dunn's post hoc test for multiple comparisons were used to compare mesenchymal and epithelial data from non-smokers, smokers and patients with COPD. Data from parametric analysis were expressed as mean (SE). Analysis of data from the in vitro mechanistic experiments in differentiated HBECs from non-smoker patients was performed by the Student t test or one-way or two-way analysis of variance followed by the Bonferroni post hoc test. Significance was accepted when p<0.05. Full details are given in the online supplement.
Results
EMT cell markers are increased in primary bronchial epithelial cells from smokers and patients with COPD
Primary HBECs were isolated from small bronchi of non-smokers, smokers and patients with COPD from uninvolved lung tissue during lobectomy/wedge resection for malignant lesions. The clinical characteristics of the patients are shown in table 1.
Clinical features of patients
Under basal conditions, primary HBECs from smokers and patients with COPD showed a significant increase in mRNA and protein expression of typical mesenchymal markers α-SMA, vimentin and collagen type I (figure 1A–C). As with the mesenchymal markers, NOX4 mRNA and protein expression were also significantly upregulated in primary HBECs from smokers and patients with COPD (figure 1D). The analysis of epithelial markers revealed a significant downregulation of E-cadherin, ZO-1, KRT5 and KRT18 mRNA in patients with COPD but not in smokers, and a decrease in E-cadherin and ZO-1 protein expression in smokers and patients with COPD (figure 1E–H). In smokers and patients with COPD, phosphorylation of ERK1/2 was significantly increased, but an increase in Smad3 phosphorylation was only observed in cells from patients with COPD (figure 1I,J). The transcription factor β-catenin was downregulated in the cytoplasm and upregulated in the nucleus of epithelial cells from smokers and patients with COPD (figure 1K). Furthermore, basal cell supernatants of 24 h cell cultures of patients with COPD showed higher MMP-9 release than those of non-smokers and smokers (figure 1L).

Expression of epithelial and mesenchymal markers in primary human bronchial epithelial cells (HBECs) from non-smokers, smokers and patients with chronic obstructive pulmonary disease (COPD). Primary HBECs were isolated from small bronchi of non-smokers (n=5), smokers (n=12) and patients with COPD (n=15) and total RNA and protein were obtained as described in the Methods section. mRNA and protein expression was determined by real-time PCR and protein array Zeptosens technology, respectively, with appropriate primers and specific antibodies. Basal expression of mRNA transcripts or protein of the mesenchymal markers (A) α smooth muscle actin, (B) vimentin, (C) collagen type I (Col type I) or (D) NOX4, as well as basal expression of the epithelial markers (E) E-cadherin, (F) ZO-1, (G) KRT5 or (H) KRT18 are shown. (I, J) Basal protein expression of phospho-Smad3 and phospho-ERK1/2. Data are expressed as the ratio to GAPDH for mRNA levels and to β-actin for protein levels and normalised to the non-smoker group. Phospho-Smad3 and phospho-ERK1/2 protein levels were expressed as the ratio to total ERK1/2 and Smad3, respectively, and normalised to the non-smoker group. (K) Representative images of western blot for cytoplasm and nuclear β-catenin with corresponding internal controls β-actin and lamin A. (L) Levels of matrix metalloproteinase (MMP)-9 were quantified by ELISA in primary culture cells following 24 h of cultures. Data are presented as box and whisker plots with median, IQR and minimum and maximum values. p exact values were obtained by the Kruskal–Wallis test and Dunn's post hoc test.
Phase contrast images showed that cells from smokers and patients with COPD had approximately 25% of cells with a spindle-shaped appearance characteristic of mesenchymal cells (figure 2). Expression of the tight junction (TJ) proteins E-cadherin and ZO-1 was visualised by immunofluorescence as a continuous line of fluorescence around the margin of the cells, and F-actin fibres were distributed at the margins of cells, between cell contacts in the non-smoker group. In contrast, cells from smokers and patients with COPD showed a reduction in E-cadherin and ZO-1 fluorescence intensity with sporadic and diffused expression. F-actin in cells from smokers and patients with COPD was homogeneously distributed as fibres in the cytoplasm. The mesenchymal markers collagen type I and vimentin were found to be upregulated in cells from smokers and patients with COPD and almost absent in cells from non-smokers (figure 2). The epithelial marker KRT5 was positively expressed in all HBECs from non-smokers, smokers and patients with COPD, although the intensity was lower in primary cultures from smokers and patients with COPD. Furthermore, positive KRT5 cells in smokers and patients with COPD were also positive for mesenchymal collagen I and vimentin positive cells (figure 2).
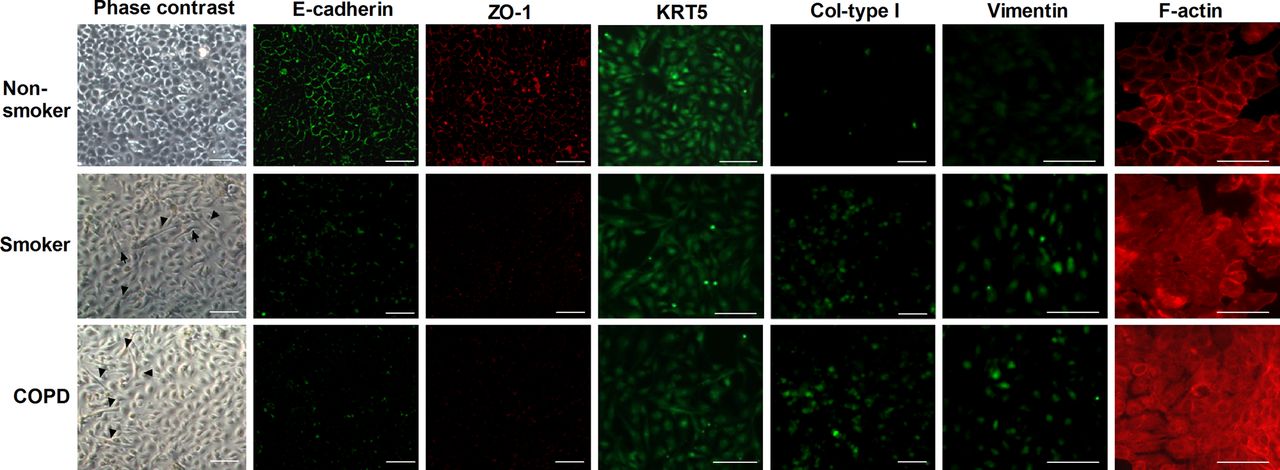
Small bronchial pieces from non-smokers (n=5), smokers (n=6) and patients with chronic obstructive pulmonary disease (COPD) (n=6) were placed on culture plates and bronchial epithelial cells grew around the explants. When the cells reached around 100% of confluence, phase contrast images and immunofluorescence staining of E-cadherin, ZO-1, KRT5 collagen type I (Col-type I) and vimentin were taken. F-actin fluorescent images were analysed with phalloidin-tetramethylrhodamine isothiocyanate. Representative images are shown (scale bar 20 µm). Black arrows show epithelial cells with spindle-shaped appearance. This figure is only reproduced in colour in the online version.
Immunohistochemical analysis of small airways revealed that α-SMA was immunostained in the submucosa of bronchi but not in bronchial epithelial cells from non-smokers (figure 3A, panel A). However, in both smokers and patients with COPD, α-SMA was markedly immunostained with a peribronchiolar location. Furthermore, α-SMA immunostaining was observed within the bronchial epithelium (figure 3A, panels F and K, black arrows). Vimentin immunostaining was absent in bronchial epithelial cells from non-smoker patients (figure 3A, panel B) but, in both smokers and patients with COPD, vimentin was located in basal epithelial cells (figure 3A, panels G and L). Collagen type I was increased in airway sections from smokers and patients with COPD, which correlates with the extracellular matrix deposition found in peribronchiolar fibrosis.3 NOX4 was weakly expressed in small lung sections from non-smokers, differing from smokers and patients with COPD who expressed NOX4 on bronchial epithelial cells as well as in bronchial infiltrated leucocytes (figure 3A, panels I and N). Epithelial ZO-1 and E-cadherin were markedly expressed in the bronchial epithelium of non-smokers (figure 3B, panels A and B), with lower expression in the bronchial epithelium of smokers and patients with COPD (figure 3B, panels D–H). Composite scores of immunohistochemistry showed higher values for α-SMA, vimentin, collagen type I and NOX4 in bronchial epithelial cells of smokers and patients with COPD compared with non-smoker patients and lower values for E-cadherin and ZO-1 (figure 3C).
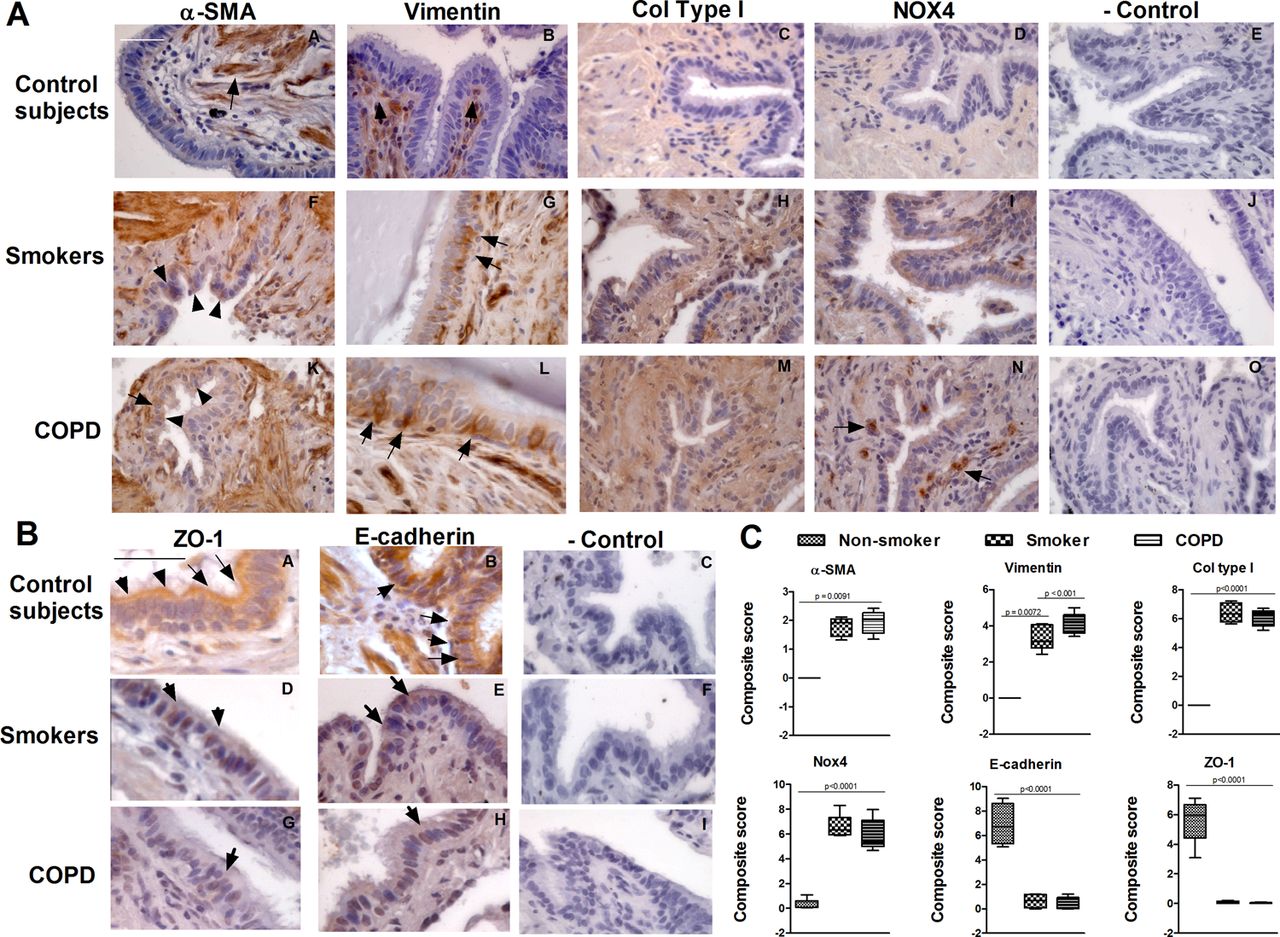
Immunohistochemistry of small airways. Small bronchial sections from non-smokers (n=4), smokers (n=10) and patients with chronic obstructive pulmonary disease (COPD) (n=10) were immunostained for (A) α smooth muscle actin (α-SMA), vimentin, collagen type I (Col type I), NOX4 (brown) and (B) epithelial ZO-1 and E-cadherin markers (brown). Black arrows show the main distribution of the positive immunostaining regions of the small bronchi. Representative immunohistochemistry images are shown (scale bar 45 µm). The control IgG isotype always gave a negative signal. (C) Box plot representing the composite score of α-SMA, vimentin, Col type I, NOX4, ZO-1 and E-cadherin markers across the bronchial epithelium in 10 slices per patient. Data are presented as box and whisker plots with median, IQR and minimum and maximum values. p exact values were obtained by the Kruskal–Wallis test and Dunn's post hoc test. This figure is only reproduced in colour in the online version.
EMT is induced by CSE in differentiated HBECs
To explore whether cigarette smoke may directly induce EMT in bronchial epithelial cells, we used an in vitro model of differentiated HBECs from non-smoker patients. CSE dose-dependently increased mesenchymal markers α-SMA and collagen type I, reaching a peak value at a concentration of 2.5% following 72 h of exposure (figure 4A; p<0.05). We therefore selected these time and concentration conditions of CSE to evaluate EMT markers in future experiments. In this respect, CSE at a concentration of 2.5% significantly increased mRNA transcripts and protein expression for the mesenchymal markers α-SMA, vimentin and collagen type I (figure 4B,C) while the epithelial markers E-cadherin, ZO-1, KRT5 and KRT18 were significantly downregulated (figure 4D,E). According to the loss of expression of TJ proteins, CSE (0.5–5%) dose-dependently decreased TEER (figure 4F) as a marker of loss of epithelial cell barrier. Furthermore, differentiated HBECs exposed to CSE adopted a flattened and elongated morphology characteristic of a myofibroblast-like phenotype. CSE-induced EMT markers were sustained for almost 7 days after the washout of CSE, which reflects the irreversibility of the EMT process (see figure C in online supplement).

Cigarette smoke extract (CSE) induces epithelial to mesenchymal transition in differentiated human bronchial epithelial cells (HBECs). Differentiated HBECs were treated with 0.5–5% CSE or vehicle for 72 h and total RNA or cell lysates were obtained for real-time PCR (A, B, D) or protein array Zeptosens (C, E). CSE induced (A, B) mRNA transcripts and (C) protein expression of mesenchymal markers α smooth muscle actin, vimentin and collagen type I (Col type I). CSE significantly reduced (D) mRNA transcripts and (E) protein expression of epithelial markers E-cadherin, ZO-1, KRT5 and KRT18. Data are expressed as the ratio to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for mRNA levels and to β-actin for protein levels and normalised to the solvent control group. (F) Development of transepithelial electrical resistance (TEER) in differentiated HBECs. TEER was measured after 72 h of CSE (0.5–5%) exposure. Results are expressed as mean (SE) of four independent experiments (one cell non-smoker population) per condition. Data analysed by one-way ANOVA followed by post hoc Bonferroni test (A) or Student t test (B–E). *p<0.05 vs solvent controls.
CSE-induced EMT is partially mediated by the increase of ROS and downregulation of cAMP
Preincubation of differentiated HBECs with the antioxidants N-acetyl-ι-cysteine (NAC) and apocynin (APO) inhibited the increase of α-SMA, vimentin and collagen type I mRNA and protein expression as well as the downregulation of the epithelial markers E-cadherin, ZO-1, KRT5 and KRT18 induced by CSE (figure 5A–D). As observed for the antioxidants, the analogue of cAMP (dbcAMP) significantly inhibited the increase in mRNA and protein expression of the mesenchymal markers α-SMA, vimentin and collagen type I as well as the downregulation of the epithelial markers E-cadherin, ZO-1, KRT5 and KRT18 following CSE exposure (figure 5A–D). Phase contrast microscopic analysis of the differentiated HBEC culture surface and analysis of H&E-stained slide sections of multilayered HBECs showed that the cells underwent a morphological change from a cobblestone-like cell morphology to an elongated and spindle-like morphology, and reduced their cell–cell contact secondary to CSE exposure (figure 5E).

Cigarette smoke extract (CSE)-induced epithelial to mesenchymal transition is partially mediated by intracellular reactive oxygen species and inhibited by adenosine cyclic monophosphate (cAMP). Differentiated human bronchial epithelial cells (HBECs) were incubated with the antioxidants N-acetyl-ι-cysteine (NAC; 1 mM) or apocynin (APO; 100 µM), or with the cAMP analogue dibutyril cAMP (dbcAMP; 1 mM) for 30 min before CSE (2.5%) stimulation for 72 h. (A, B) Total RNA and (C, D) protein were isolated for real-time PCR and protein array Zeptosens analysis, respectively. CSE-induced upregulation of (A) mRNA and (C) protein expression of mesenchymal markers α smooth muscle actin, vimentin and collagen type I (Col type I) was inhibited by NAC, APO and dbcAMP. CSE-induced downregulation of (B) mRNA and (D) protein expression of epithelial markers E-cadherin, ZO-1, KRT5 and KRT18 was inhibited by NAC, APO and dbcAMP. Data are expressed as the ratio to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for mRNA levels and to β-actin for protein levels and normalised to the solvent control group. Results are expressed as mean (SE) of n=4 (four cell non-smoker population) experiments per condition. Data analysed by two-way ANOVA followed by post hoc Bonferroni tests. *p<0.05 vs solvent controls; #p<0.05 vs CSE. (E) Morphological changes in differentiated HBECs after 72 h of exposure to 2.5% CSE showed a mesenchymal morphology with reduced cell–cell contact as assessed by phase contrast light microscopy (apical surface) and H&E section histology. Morphological changes were inhibited by NAC and dbcAMP. Representative images are shown (scale bar 40 µm). This figure is only reproduced in colour in the online version.
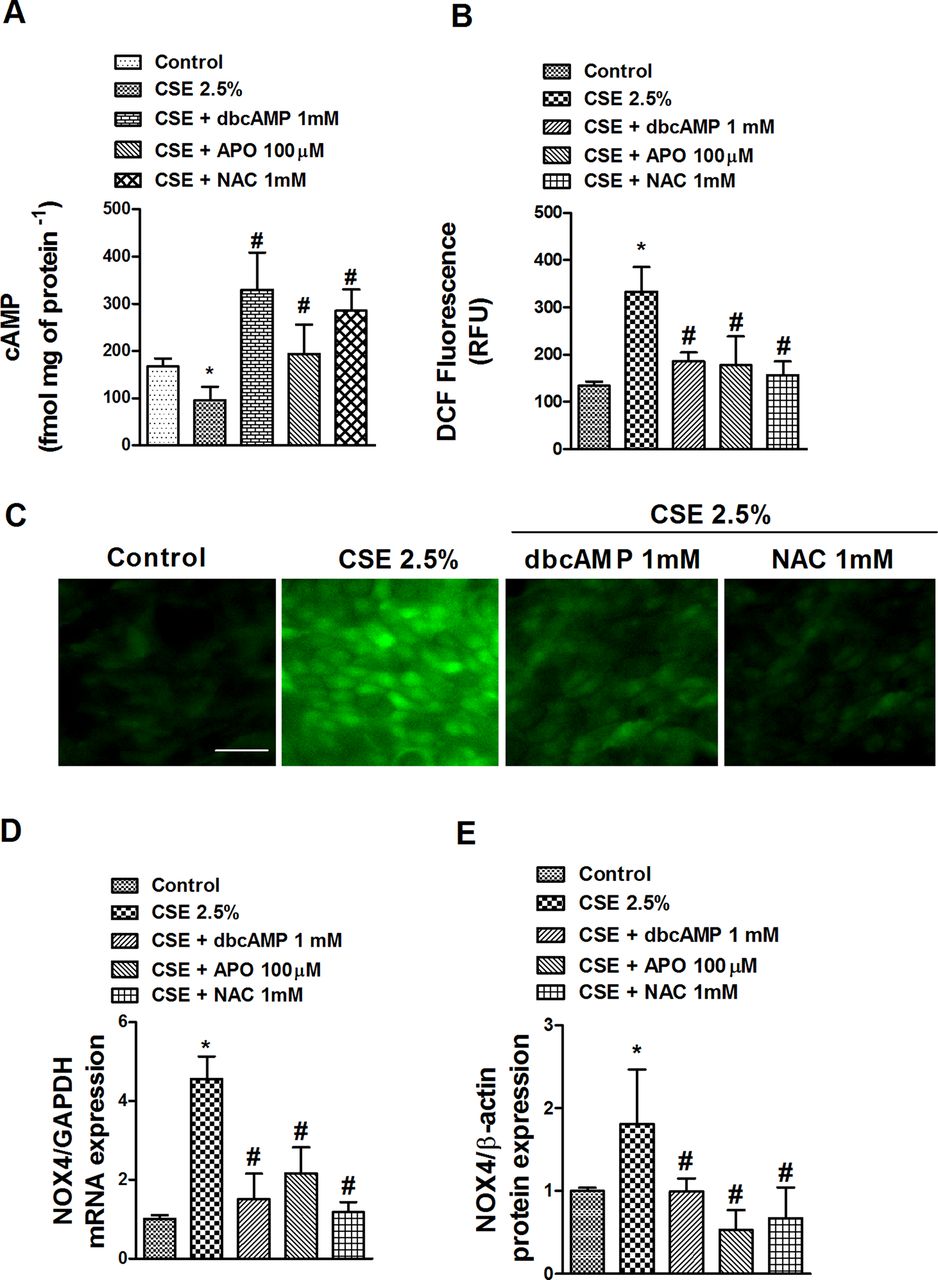
Interestingly, CSE 2.5% significantly decreased the intracellular cAMP content after 72 h, which was significantly inhibited by the antioxidants NAC and APO and the positive control dbcAMP (figure 6A), suggesting a role for ROS in cAMP downregulation. In this context, CSE 2.5% increased intracellular ROS after 30 min of exposure (figure 6B,C) and sustained this increase for the 72 h period (data not shown). The increase in ROS was accompanied by an increase in mRNA and protein expression of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase unit NOX4 after 72 h of exposure (figures 6D,E). Both ROS production and NOX4 expression induced by CSE were inhibited by dbcAMP, NAC and APO in differentiated HBECs (figure 6B–E).

Cigarette smoke extract (CSE) reduces adenosine cyclic monophosphate (cAMP) intracellular levels and increases reactive oxygen species (ROS) and NOX4 expression in differentiated human bronchial epithelial cells (HBECs). Differentiated HBECs were incubated with the antioxidants N-acetyl-ι-cysteine (NAC; 1 mM) or apocynin (APO; 100 µM), or with the cAMP analogue dibutyril cAMP (dbcAMP; 1 mM) for 30 min before CSE (2.5%) stimulation for 72 h (A, D, E) or 30 min (B, C). (A) CSE-induced loss of intracellular cAMP was inhibited by NAC, APO and positive control dbcAMP. (B) ROS were determined by means of DCF fluorescence intensity after CSE stimulation in the presence or absence of NAC, APO or dbcAMP. (C) Representative images of positive DCF fluorescence in differentiated HBECs (scale bar 20 µm). (D, E) CSE induced mRNA and protein expression of NOX4 which were significantly inhibited by dbcAMP, APO and NAC (D, E). Data are expressed as the ratio to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for mRNA levels and to β-actin for protein levels and normalised to the solvent control group. Results are expressed as means (SE) of n=4 (four cell non-smoker population) experiments per condition. Data analysed by two-way ANOVA followed by post hoc Bonferroni tests. *p<0.05 vs solvent controls; #p<0.05 vs CSE. RFU, relative fluorescence units. This figure is only reproduced in colour in the online version.
TGF-β1, ERK1/2 and Smad3 mediate CSE-induced EMT in differentiated HBECs
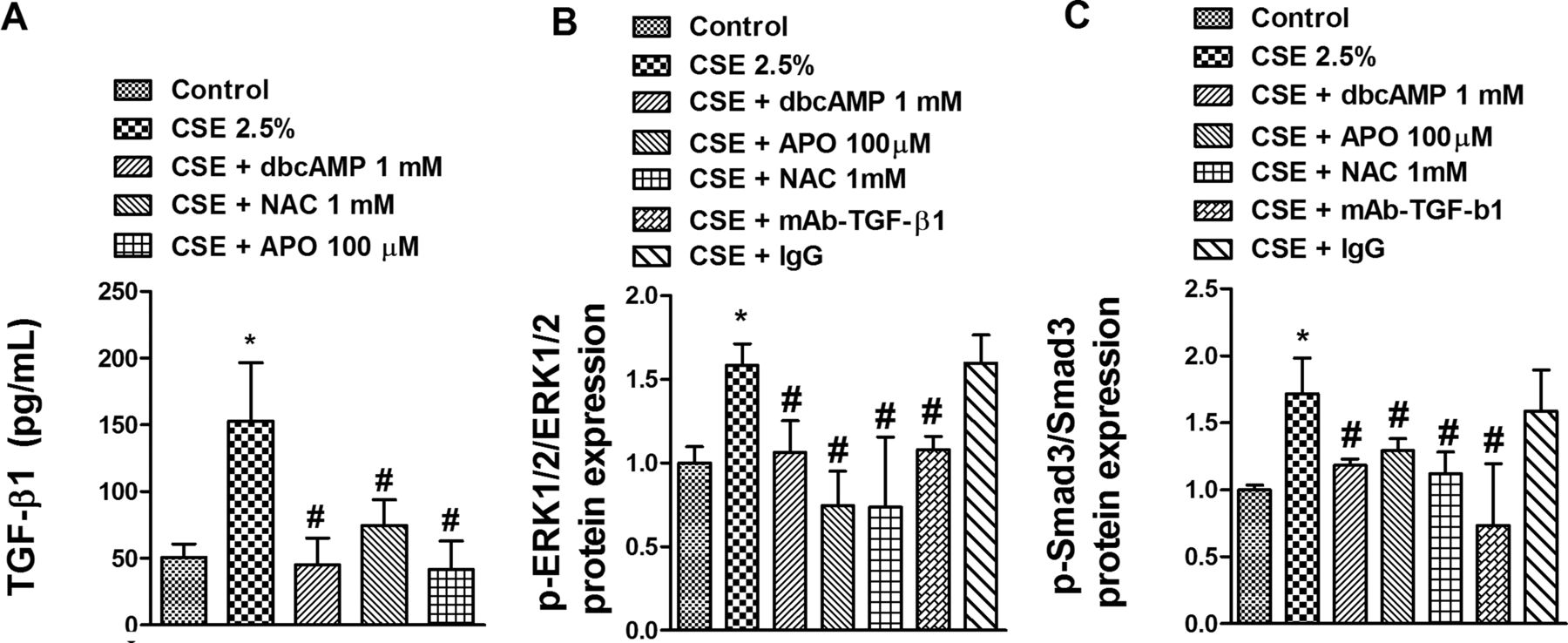
Stimulation with CSE 2.5% for 72 h significantly increased by approximately 2.5-fold the release of the active form of TGF-β1 (figure 7A). Preincubation of differentiated HBECs with dbcAMP, NAC or APO completely suppressed the release of TGF-β1 (figure 7A), suggesting a role for ROS and cAMP in mediating CSE-induced TGF-β1 release. In other experiments, CSE 2.5% stimulation increased the phosphorylation of ERK1/2 and Smad3 which was significantly inhibited by anti-TGF-β1 antibody (mAb-TGF-β1) (figure 7B,C) suggesting an autocrine role for TGF-β1. In parallel, preincubation of differentiated HBECs with dbcAMP, NAC or APO also inhibited CSE-induced ERK1/2 and Smad3 phosphorylation.

Cigarette smoke extract (CSE) increases secretion of transforming growth factor β1 (TGF-β1) and phosphorylation of ERK1/2 and Smad3 in differentiated human bronchial epithelial cells (HBECs). Differentiated HBECs were incubated with the antioxidants N-acetyl-ι-cysteine (NAC; 1 mM) or apocynin (APO; 100 µM), or with the cyclic monophosphate (cAMP) analogue dibutyril cAMP (dbcAMP; 1 mM) for 30 min before CSE (2.5%) stimulation for 72 h. In panels B and C an anti-TGF-β1 antibody (mAb-TGF-β1; 4 µg/ml) and its negative control non-immune IgG were added for 30 min before CSE 2.5% stimulation for 72 h. Cell culture supernatant and protein content were extracted to quantify (A) TGF-β1 release, (B) ERK1/2 phosphorylation and (C) Smad3 phosphorylation. (B, C) Phospho-Smad3 and phospho-ERK1/2 protein levels were expressed as the ratio to total ERK1/2 and Smad3, respectively, and normalised to the solvent control group. Results are expressed as mean (SE) of n=4 (four cell non-smoker population) experiments per condition. Data analysed by two-way ANOVA followed by post hoc Bonferroni tests. *p<0.05 vs solvent controls; #p<0.05 vs CSE.
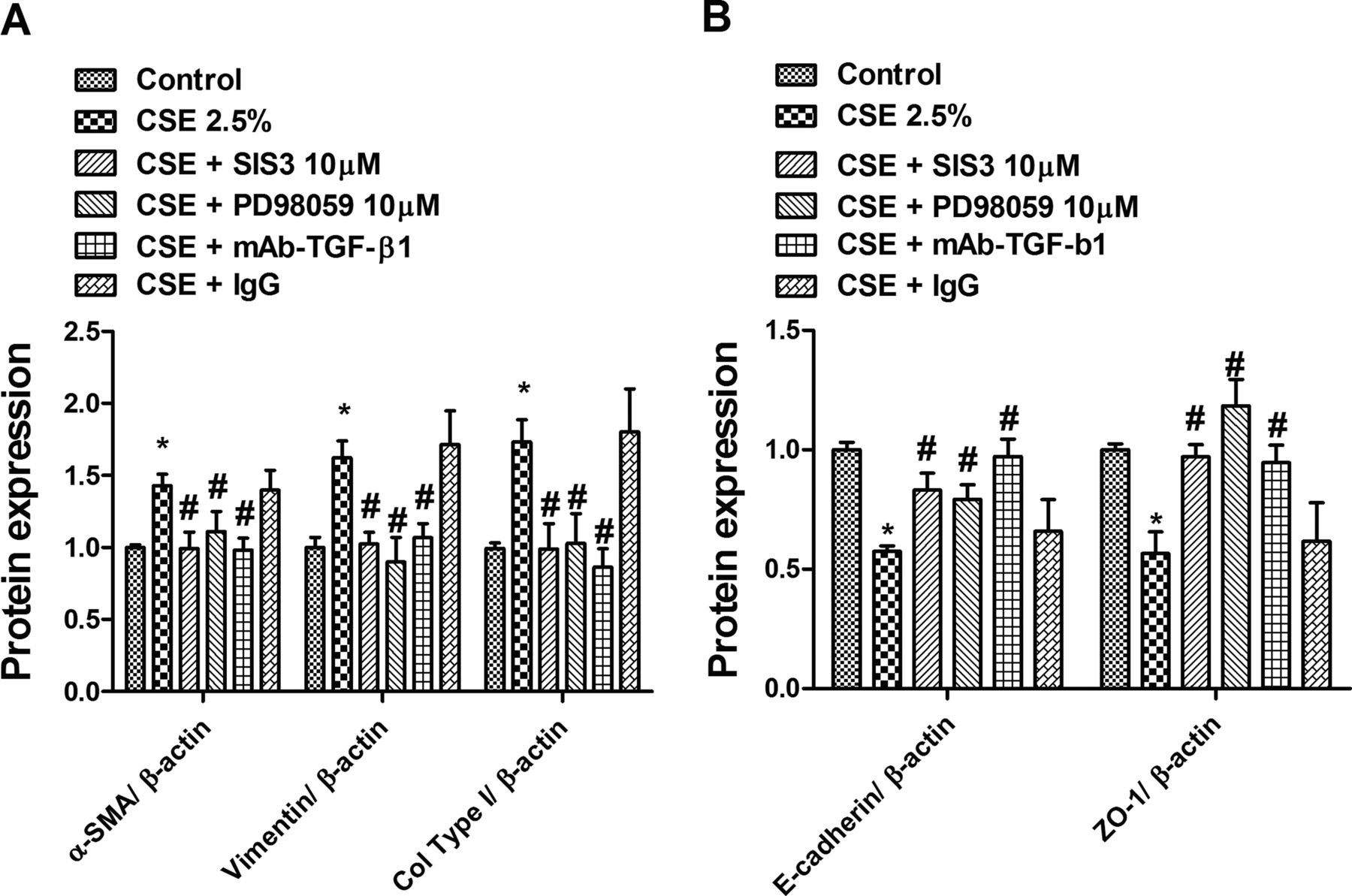
Finally, to analyse the autocrine effect of TGF-β1 and its downstream pathways ERK1/2 and Smad3 in the CSE-induced EMT process, we preincubated cells with mAb-TGF-β1, the ERK1/2 antagonist PD98058 or with the Smad3 inhibitor SIS3. All of them nearly suppressed CSE-induced upregulation of the mesenchymal markers α-SMA, vimentin and collagen type I (figure 8A), as well as downregulation of the epithelial markers E-cadherin and ZO-1 (figure 8B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cigarette smoke extract (CSE)-induced epithelial to mesenchymal transition is partially mediated by an autocrine transforming growth factor β1 (TGF-β1) effect and its downstream signalling phospho-ERK1/2 and phospho-Smad3. Differentiated human bronchial epithelial cells (HBECs) were incubated with the Smad3 inhibitor SIS3 (10 µM), the ERK1/2 inhibitor PD98059 (10 µM), the anti-TGF-β1 antibody (mAb-TGF-β1; 4 µg/ml) or its negative control non-immune IgG for 30 min before CSE (2.5%) stimulation for 72 h. After incubation periods, differentiated HBECs were lysed and protein array Zeptosens performed. (A) CSE-induced upregulation of α smooth muscle actin, vimentin and collagen type I (Col type I) mesenchymal markers was inhibited by SIS3, PD98059 and mAb-TGF-β1. (B) CSE-induced downregulation of E-cadherin and ZO-1 epithelial markers was inhibited by SIS3, PD98059 and mAb-TGF-β1. (A, B) Data are expressed as the ratio to β-actin for protein levels and normalised to the solvent control group. Results are expressed as means (SE) of n=4 (four cell non-smoker population) experiments per condition. Data analysed by two-way ANOVA followed by post hoc Bonferroni tests. *p<0.05 vs solvent controls; #p<0.05 vs CSE.
Discussion
The main and novel results of the present study are that (1) primary HBECs of small bronchi from smokers and patients with COPD show an increase in the typical EMT expression profile and (2) in vitro stimulation of differentiated HBECs with CSE promotes EMT through the activation of intracellular ROS, the release of TGF-β1, the phosphorylation of ERK1/2 and Smad3 as well as by the downregulation of cAMP. These new findings suggest that EMT may be induced directly by cigarette smoke, and that EMT is present in the small bronchi of smokers and patients with COPD, which may potentially contribute to the thickening of the wall of the small airways caused by fibrosis.
In the airways EMT has been implicated in several lung fibrotic disorders.14 However, the hypothesis that lung bronchial fibroblasts may originate directly from HBECs as a result of EMT remains to be elucidated. In this respect, recently investigated in vivo biopsy specimens and ex vivo primary HBEC cultures from lung allograft recipients showed evidence of EMT.18 In addition, Hackett and colleagues observed EMT in differentiated HBECs in response to TGF-β1.8 However, there is little knowledge regarding the potential for EMT to occur in COPD. In this regard, Sohal and colleagues recently observed that large airway biopsies from smokers and patients with COPD potentially undergo EMT.13 ,19 However, the findings provided by these studies were limited since the mesenchymal markers analysed (MMP-9, vimentin and S100A4) are also expressed in other cell types such as leucocytes and endothelial cells.20 ,21 Furthermore, Sohal and colleagues studied EMT in large airways. Since bronchial remodelling in COPD, such as peribronchiolar fibrosis, is mainly located in small conducting airways (bronchioles <2 mm in diameter), and considering that large and small airways differ in their anatomical and pathophysiological characteristics,22 previous findings need to be corroborated in small bronchi. In the present study we analysed the expression of mesenchymal markers broadly referenced in EMT studies such as the cytoskeleton proteins α-SMA and vimentin as well as the extracellular matrix protein collagen type I,7 being α-SMA and collagen type I-specific for the fibroblast/myofibroblast phenotype.7 ,23 Since EMT is characterised by the loss of epithelial cobblestone or cuboidal morphology and a marked reduction in epithelial E-cadherin, ZO-1 and cytokeratin expression, we also analysed the loss of expression of these epithelial markers as indicators of the EMT process. In this regard, positive KRT5 cells in cells from smokers and patients with COPD were also positive for mesenchymal collagen I and vimentin positive cells (figure 2). During the process of EMT some epithelial and mesenchymal cell markers are known to be co-expressed within individual cells,24 which might explain why we have observed positive cells for both markers in HBECs from smokers and patients with COPD.
To our knowledge, this is the first report showing α-SMA in basal epithelial cells as a marker of human myofibroblasts in the small bronchi of smokers and patients with COPD. In this respect, recent work in a model of α-SMA-Cre/R26R mice of bleomycin-induced pulmonary fibrosis also showed upregulation of α-SMA in basal bronchial epithelial cells, supporting an in vivo EMT process in the bronchial epithelium.25 Vimentin was also immunostained in basal epithelial cells from smokers and patients with COPD, as previously observed in large airways,26 and the expression of collagen type I was also upregulated in small bronchi of smokers and patients with COPD.
The epithelial markers E-cadherin and ZO-1 proteins were found to be downregulated in primary HBECs and the epithelium of small bronchi from smokers and patients with COPD. However, E-cadherin and ZO-1 mRNA expression was not decreased in smokers. These contradictory findings may be explained by E-cadherin and ZO-1 mRNA and protein expression being regulated at different levels. Thus, cell membrane E-cadherin and ZO-1 proteins as part of TJs between epithelial cells are degraded by different metalloproteinases released by the effect of oxidant agents such as cigarette smoke. In fact, it has been observed previously that ZO-1 is one means by which occludin is anchored to the cytoskeleton to maintain TJ. In the presence of oxidative stress, ZO-1:occludin association and E-cadherin is decreased and causes the diffuse cytoplasmic redistribution and degradation by metalloproteinases27 ,28 excluding transcriptional mechanisms. Another independent system is governed by transcriptional mechanisms in which the effect of some remodelling factors such as TGF-β1 may reduce mRNA transcription of ZO-1 and E-cadherin.29 The differences between mRNA and protein expression of ZO-1 and E-cadherin observed between smokers and patients with COPD could therefore be due to a predominant role of protein degradation in bronchial epithelial cells from patients with COPD because of a high level of metalloproteinase expression in their lung tissues.30 Accordingly, we found higher basal levels of MMP-9 in cells from patients with COPD than in smokers.
Despite the novelty of our results, some limitations should be highlighted. In particular, both the primary cell culture and lung tissue analysed could be criticised. The loss of cell phenotype in prolonged culture is well described and could modify the results in primary HBECs. However, our primary ex vivo cell culture experiments were performed in HBECs that were grown around the explants of small bronchi until they reached approximately 100% of confluence, forming an epithelial cell layer (without passages and without trypsinisation) on KRT5 positive cells in order to prevent phenotypic changes, as previously outlined.18 On the other hand, the lung tissues studied were from patients undergoing lung surgery for lung carcinoma. Since EMT has been well characterised in epithelial lung malignant disease,31 it could be argued that the EMT expression profile is due to spread of tumour lesions. However, samples of distal lung, located as far away as possible from the tumour, were carefully checked histologically to exclude the presence of tumour.
Another limitation could be related to the fact that the observed increase in the expression of mesenchymal markers in the epithelium is related to the decline in lung function while increased mesenchymal markers are also observed in smokers with normal lung function. This apparent contradiction may be explained by the fact that remodelling is a set of processes affecting whole airways which differ between smokers and patients with COPD. Patients who are smokers show less goblet cell and submucosal gland hyperplasia than patients with COPD,2 absence of loss of alveolar wall, less vascular remodelling32 and less airway wall thickening and smooth muscle deposition around bronchi,33 all contributing to lung function. Thus, although mesenchymal markers were upregulated in smokers with normal lung function, this may be considered as an initial step in small bronchial remodelling contributing to lung function over time together with the other remodelling processes.
Next we analysed possible mediators involved in the EMT process. A hypothesis generated from animal models is that cigarette smoke drives small airways remodelling by induction of profibrotic growth factors (eg, connective tissue growth factor, platelet-derived growth factor-β and TGF-β1) from the airway wall.34 These growth factors, which have also been implicated as inducers of EMT, could contribute to peribronchiolar fibrosis. Supporting this hypothesis, several in vitro studies have shown that components of cigarette smoke such as nicotine and benzo(a)pyrene induce EMT in immortalised airway epithelial cells.35 ,36 In this study we first showed evidence that differentiated HBECs undergo EMT in response to cigarette smoke, as demonstrated by specific markers and phenotypic changes. Cigarette smoke contains a high number of ROS that may activate latent TGF-β1 release.37 Previous data support the hypothesis that cigarette smoke may release the active form of TGF-β1, activating its downstream signalling Smad2/3 cascade in bronchial rat explants to promote small bronchi remodelling.37 In addition, both cigarette smoke and TGF-β1 also promote the activation of ERK1/2 signalling in HBECs,38 suggesting a role for Smad3 and ERK1/2 phosphorylation in cigarette smoke-induced EMT. In this study we add new evidence to the mechanism by which CSE increases the release of the active form of TGF-β1, the phosphorylation of Smad3 and ERK1/2, thus promoting the EMT process in differentiated HBECs. The release of TGF-β1 and the downstream phosphorylation of Smad3 and ERK1/2 were activated by ROS generated by CSE since the antioxidants NAC and APO—as well as blocking TGF-β1 with mAb-TGF-β1—almost suppressed CSE-induced TGF-β1 signalling and EMT. These in vitro findings are of clinical relevance since the phosphorylated forms of ERK1/2 and Smad3 were upregulated in primary HBECs from smokers and patients with COPD.
The importance of oxidant-mediated TGF-β release as well as ROS in fibrotic diseases has been previously documented.39 In fact, myofibroblast transformation in lung fibrotic disorders following TGF-β1 stimulation is partially mediated by the NADPH oxidase unit NOX4.39 The plasmatic membrane unit NOX4 does not require interaction and activation by cytosolic regulatory subunits, so its activation is directly related to its expression.40 In this study, CSE increased intracellular ROS as well as NOX4 expression in differentiated HBECs. Furthermore, NOX4 expression was upregulated in primary HBECs from smokers and patients with COPD and located on bronchial epithelial cells and leucocyte infiltrates in human small bronchi, confirming activation of oxidative stress in the tissues studied.
Considering the established role for cAMP as an inhibitor of ROS in epithelial cells as well as the subsequent EMT process,10 we explored whether cAMP could modulate CSE-induced EMT in differentiated HBECs. In our in vitro model CSE induced the loss of cAMP, probably by an increase in the degrading cAMP phosphodiesterase 4 (PDE4) activity, as we recently reported in HBECs.15 The addition of dbcAMP as an analogue of cAMP reduced ROS, TGF-β1 secretion and their downstream signalling phospho-ERK1/2 and phospho-Smad3 as well as the EMT process. Modulation of cAMP levels could therefore be an appropriate therapy to attenuate EMT and small bronchi remodelling in patients with COPD.
A rational sequence of mechanistic events by which CSE induces EMT in HBECs therefore includes activation of the NADPH oxidase system and upregulation of the NADPH oxidase component NOX4 to produce intracellular ROS. Thus, ROS decrease cAMP levels through the increase in PDE4 activity. In a subsequent event, the increase in intracellular ROS together with the reduction in cAMP contributes to the release of TGF-β1. TGF-β1 activates Smad3 and ERK1/2, contributing to the increase in EMT.
In summary, our data provide evidence of an active EMT process in the small bronchi of smokers and patients with COPD which may potentially contribute to the thickening of the wall of the small airways. In vitro studies show that cigarette smoke may induce EMT, modulating the TGF-β1 pathway as well as ROS and cAMP levels. Future treatments directed at these molecular pathways could potentially attenuate in vivo EMT and small bronchial remodelling of patients with COPD.
Acknowledgments
We thank G Juan and E Guijarro, Respiratory and Surgery Units, Hospital General, Valencia, Spain for facilitating clinical data and tissue collection.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
JM and TP contributed equally.
-
Contributors Concept and design: JM, JC. Acquisition of data: JM, TP, AS. Analysis and interpretation of data: JM, JC, AS, TP. Drafting the article or revising it critically for important intellectual content: JC, JM. All authors approved the final version of the manuscript.
-
Funding This work was supported by grants SAF2011-26443 (JC), FIS CP11/00293(JM), CIBERES (CB06/06/0027), ADE10/00020 (Spanish Government), ACIF/2010/114 (TP) and research grants from Regional Government (Prometeo/2008/045, ‘Generalitat Valenciana’. Support from the CENIT programme (Spanish Government) was obtained.
-
Competing interests None.
-
Ethics approval This study has been approved by the ethics committee of the University General Hospital of Valencia, Spain.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer-reviewed.