Article Text

Abstract
Background Airborne microbial products have been reported to promote immune responses that suppress asthma, yet how these beneficial effects take place remains controversial and poorly understood.
Methods We exposed mice to the bacterium Escherichia coli and subsequently induced allergic airway inflammation through sensitization and intranasal challenge with ovalbumin.
Results Pulmonary exposure to the bacterium Escherichia coli leads to a suppression of allergic airway inflammation. This immune modulation was neither mediated by the induction of a T helper 1 (Th1) response nor regulatory T cells; however, it was dependent on Toll-like receptor 4 (TLR4) but did not involve TLR desensitisation. Dendritic cell migration to the draining lymph nodes and activation of T cells was unaffected by prior exposure to E.coli, while dendritic cells in the lung displayed a less activated phenotype and had impaired antigen presentation capacity. Consequently, in situ Th2 cytokine production was abrogated. The suppression of airway hyper-responsiveness was mediated through the recruitment of gd T cells; however, the suppression of dendritic cells and T cells was mediated through a distinct mechanism that could not be overcome by the local administration of activated dendritic cells, or by the in vivo administration of tumour necrosis factor a.
Conclusion Our data reveal a localized immunoregulatory pathway that acts to protect the airways from allergic inflammation.
- Mouse
- asthma
- dendritic cell
- macrophage
- bacteria
- allergic lung disease
- lymphocyte biology
- macrophage biology
Statistics from Altmetric.com
- Mouse
- asthma
- dendritic cell
- macrophage
- bacteria
- allergic lung disease
- lymphocyte biology
- macrophage biology
Key messages
What is the key question?
Exposure to environmental bacteria can influence the development of allergy; what is the cellular mechanism through which this occurs?
What is the bottom line?
Exposure to bacteria in the lung can dysregulate antigen-presenting cell activity and protect against the development of allergic inflammation.
Why read on?
Find out about novel pathways that regulate inflammation in the lung.
Introduction
Mucosal tissues, such as the lung, are constantly exposed to both innocuous and pathogenic environmental antigens. Cells situated in close proximity to these surfaces play a central role in orchestrating immune responses upon antigen encounter, deciding between induction of tolerance or initiation of an effector immune response. This process needs to be tightly regulated, as disruption of homeostasis can lead to the development of detrimental immune responses such as allergy and asthma. Allergic asthma is driven by an inflammatory response against normally harmless environmental antigens in the respiratory tract, and is characterised by eosinophilia, goblet cell hyperplasia, bronchial hyper-responsiveness and airway tissue remodelling.1 Dendritic cells (DCs) play a crucial role in the initiation of an asthma response; following antigen uptake, they migrate to the draining lymph nodes (LNs), where they drive effector T helper 2 (Th2) cell development.2 Importantly, DCs have also been shown to be required not only for the initiation of asthma, but also for the maintenance of inflammatory Th2 responses in the lung.3 Regulation of DC function has therefore been proposed as a possible therapeutic target for suppression of asthma.
Immune responses to airborne pathogens such as viruses and bacteria have been shown to influence the development of allergic responses in the respiratory tract4 5; in some cases suppression of asthma was observed,6 7 while in others it was exacerbated.8 In mice, treatment with microbial products such as endotoxins,9 CpG-containing oligonucleotides10 or other Toll-like receptor (TLR) ligands11–13 could inhibit the classical features of asthma such as eosinophil recruitment, interleukin 5 (IL-5) production and airway hyper-responsiveness (AHR). Microbial products have been proposed to drive suppression of asthma via the induction of counteracting Th1 or CD8 T cells as well as T regulatory cell (Treg) responses.6 14 15 Notably, there does not appear to be a single immunomodulatory mechanism, indicating that specific challenges may initiate distinct cellular pathways in order to protect the integrity of the lung. We have found a novel pathway of protection against allergic airway inflammation (AAI) in a model where exposure to the bacterium Escherichia coli leads to a profound suppression of Th2 responses, characterised by reduced AHR, decreased eosinophilia and cytokine production by T cells in the lung. The mechanism involves two pathways: suppression of AHR by recruited γδ T cells; and dampening of DC function in the lung, resulting in ineffective antigen presentation to effector T cells. As a result, antigen-specific T cells that were recruited to the airways displayed impaired cytokine production in situ. The suppressive effect caused by the bacteria was TLR4 dependent, long lasting and surprisingly mediated by the induction neither of Th1 responses nor of Tregs. Our data highlight the impact exposure to airborne bacteria can have in altering cell populations and their function in the lung, thus modulating the outcome of subsequent inflammatory responses.
Methods
Mice and pathogens
Mice were maintained specific pathogen-free at BioSupport, Zürich in isolated ventilated cages. C57BL/6 and BALB/c wild-type mice were obtained from Charles River Breeding Laboratories. All gene-deficient mice were bred at Biosupport. OT-II OVA-transgenic mice were kindly provided by Dr Federica Sallusto (IRB, Bellinzona, Switzerland). Animals used in experiments were between 8 and 12 weeks of age. All experiments were performed with permission from the Zürich Animal Ethics Committee.
Glycerol stocks of E coli DH5αpDS (DS Red) were grown overnight in Luria–Bertani (LB) medium supplemented with ampicillin; 107 colony-forming units (CFU) of E coli were resuspended in phosphate-buffered saline (PBS) and administered intranasally.
Ovalbumin-induced airway inflammation
Mice were immunised by intraperitoneal injection with 100 μg of ovalbumin (OVA, grade V; Sigma-Aldrich, Saint Louis, USA) in 200 μl of alum adjuvant (SERVA Electrophoresis GmbH, Heidelberg, Germany). Seven days later, mice were challenged by intranasal inoculation with 100 μg OVA in 50 μl of PBS. E coli were administered 3 days prior to immunisation with OVA in alum unless stated otherwise. Analysis of AAI was performed on day 3–4 after intranasal challenge. Fluorescein isothiocyanate (FITC)-labelled OVA (Molecular Probes, Basel, Switzerland) was administered at a concentration of 2 mg/ml. For challenge studies with bone marrow-derived dendritic cells (BM-DCs), cells were generated as previously described,3 pulsed for 6 h with 100 μg/ml OVA and administered intranasally in 50 μl of PBS. Unpulsed BM-DCs were administered as a control. For lipopolysaccharide (LPS) challenge studies, mice were immunised with OVA in alum and challenged with 1, 10 or 100 ng of LPS (Invivogen, San Diego, USA) 7 days later. Control mice were not treated with E coli prior to immunisation. Organ and cell isolation, flow cytometry and ELISA analysis are described in the Supplementary methods section online.
Proliferation assay and transfer of Th2 cells
CD4 T cells were purified from draining LNs or splenic single-cell suspensions by magnetic separation (MACS; Milteny Biotech, Bergisch Gladbach, Germany). For proliferation assay, cells were cultured for 72 h at 37°C with [3H]thymidine (1 μCi/well) added for the last 18 h, and total [3H]thymidine incorporation was measured. For Th2 cell differentiation, OVA-transgenic CD4 T cells from OT-II mice were cultured for 3 days at 37°C in the presence of freshly isolated splenic CD11c+ DCs, 5 μg/ml OVA-peptide323–339 (EMC Microcollections, Tübingen, Germany), 10 ng/ml recombinant IL-4 (rIL-4; BD Pharmingen, San Jose, USA) and 10 μg/ml anti-interferon γ (IFNγ) antibody (clone XMG1.2). Th2 cells were tested for IL-4 and IL-5 production by intracellular staining and flow cytometry. A total of 3×106 cells were injected intravenously into CD45.1+ recipient mice; on the same day, and for the following 3 days, mice were challenged with 100 μg of OVA in PBS.
Statistical analysis
Two independent groups (non-treated mice vs E coli-treated mice) were compared throughout the study; we assumed normal distribution of values and similar SDs in both experimental groups. Statistically significant differences between non-treated and E coli-treated mice were determined by unpaired, two-tailed Student t test. p Values <0.05 were indicative of significance and are displayed with an asterisk.
Results
Administration of E coli to the lung leads to long-term suppression of AAI
Immune responses to airborne pathogens have been shown to influence the development of allergy in the lung. In this study we sought to investigate the influence of E coli exposure in the lung on the development of AAI by treating the mice with bacteria prior to disease induction. The airway microbiota has only recently begun to be characterised16; thus, in our experiments, we utilised a well-characterised E coli strain as a model for non-pathogenic bacterial encounter in the lung. The E coli strain used does not cause lung pathology but does cause an acute influx of neutrophils into the airways shortly after administration (Supplementary figure 1).
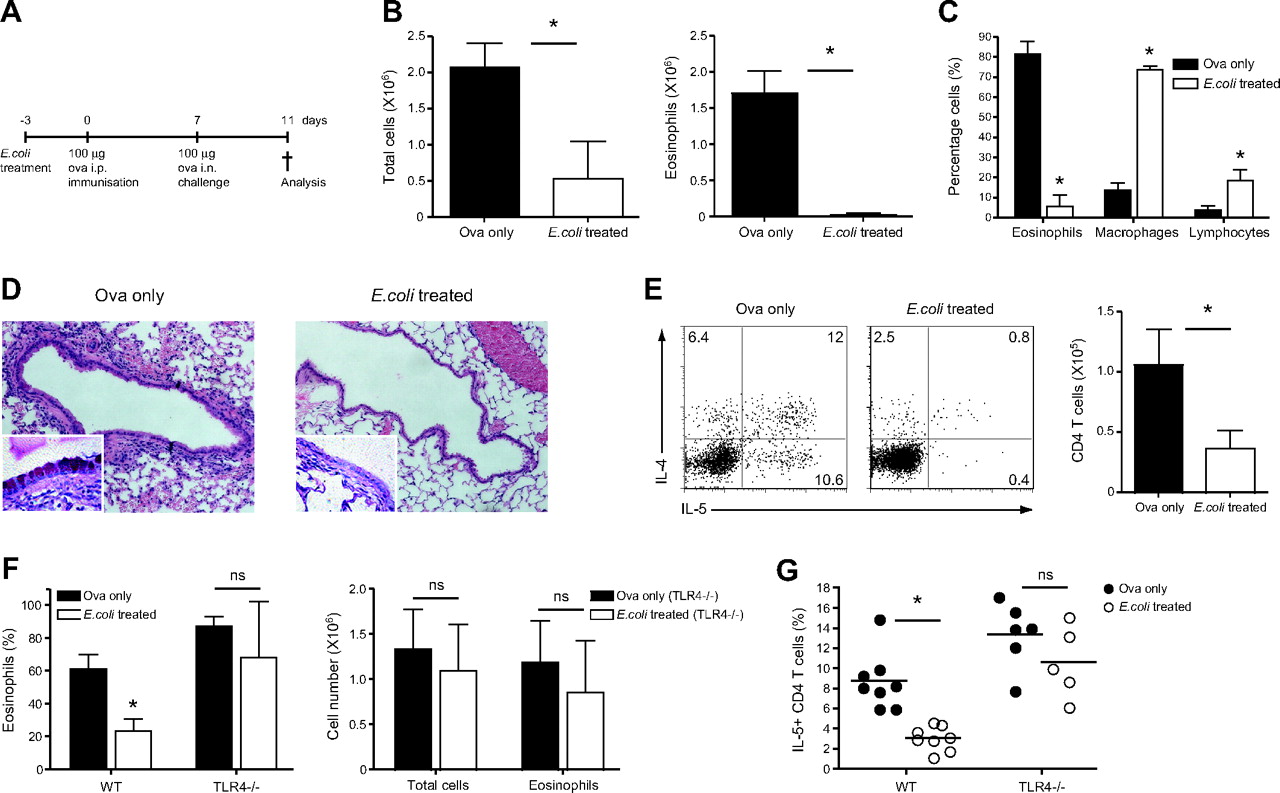
Wild-type C57BL/6 mice were treated with E coli and 3 days later mice were sensitised by intraperitoneal injection of the protein antigen OVA in alum. Mice were subsequently challenged intranasally with OVA on day 7 after immunisation (figure 1A). After 4 days, that is 2 weeks after E coli treatment, E coli- and non-treated mice were sacrificed, and the development of AAI, such as cell recruitment to the airways, was evaluated. Mice treated with E coli prior to disease induction had significantly fewer cells in the airways when compared with control mice (figure 1B, left panel). The latter had high frequencies of eosinophils in the bronchoalveolar lavage (BAL) (figure 1B,C), in contrast to the mice treated with the bacteria whose airway cellular infiltrates mostly consisted of monocytes/macrophages as determined by differential cell counts (figure 1C). Histological analysis of lung sections confirmed a decrease in cellular recruitment in E coli-treated mice; in addition, reduced goblet cell hyperplasia was observed when compared with control mice (figure 1D).

Intranasal E coli treatment prior to induction of allergic airway inflammation suppresses cardinal disease features in a Toll-like receptor 4 (TLR4)-dependent manner. C57BL/6 mice were treated with 107 colony-forming units (CFU) of E coli intranasally and 3 days later sensitised intraperitoneally with ovalbumin (OVA) in alum. Seven days later, mice were challenged intranasally with OVA in phosphate-buffered saline (PBS). Alternatively, mice were sensitised and challenged with OVA without prior inhalation of bacteria (OVA only). (A) Summary of experimental protocol. (B) On day 4 after intranasal challenge, total cell numbers in the bronchoalveolar lavage (BAL) were determined. Proportions of eosinophils, macrophages and lymphocytes were determined by differential cell counts (C), and total eosinophil numbers calculated accordingly. (D) Lungs were collected in formalin solution; histological lung sections show cellular infiltration as determined by H&E staining and goblet cell hyperplasia (insert, 100× magnification) as determined by Alcian Blue and periodic acid–Schiff (PAS) staining. (E) Cytokine production by BAL CD4 T cells (left panel). Values in FACS (fluorescence-activated cell sorting) plots represent the percentage of cells detected in each gate. Representative data from one mouse per group are shown. Total CD4 numbers were determined by flow cytometry (right panel). (F) Non-treated and E coli-treated BALB/c and TLR4-deficient mice were sensitised and challenged with OVA, and the proportion of eosinophils in the BAL was determined on day 4 after challenge (left panel). Total cell numbers in the BAL were determined, and total eosinophil numbers calculated according to cell proportions (right panel). (G) Proportion of interleukin 5 (IL-5)-producing CD4 T cells in the BAL as determined by intracellular staining. Results represent values from single animals or are presented as mean±SD. *p <0.05; statistically significant differences. Experiments were repeated ≥2 times with 4–8 mice per group. i.n., intranasal; i.p., intraperitoneal; WT, wild type.
We sought to investigate whether the observed suppression of AAI by E coli administration correlated with an impaired Th2 response. Following in vitro restimulation, IL-4 and IL-5 production by BAL CD4 T cells was largely abrogated in E coli-treated mice as compared with non-treated, OVA-sensitised mice (figure 1E). Moreover, recruitment of total CD4+ T cells was also reduced (figure 1E, right panel). E coli treatment was able to induce a long-lasting suppressive effect on the development of AAI, as a similar reduction in eosinophil recruitment and Th2 cytokine production could be observed when mice were challenged with OVA 30 days after administration of the bacteria (Supplementary figure 2).
E coli-mediated suppression is TLR4 dependent
TLR4 and its stimulation by endotoxin has been previously shown to influence the outcome of asthmatic responses.17 18 To investigate the involvement of this receptor in our model, we performed the experiment described above in TLR4-deficient mice, which were on a BALB/c genetic background. Notably a comparable suppression of AAI by E coli administration was observed in both BALB/c and C57BL/6 mice. As expected, on day 4 after challenge, wild-type BALB/c E coli-treated mice displayed lower eosinophil frequencies as compared with their non-treated counterparts. However, this reduction was not observed in TLR4-deficient mice, as both non- and E coli-treated mice had high percentages of eosinophils in the BAL (figure 1F, left). Total cell infiltration in the airways of TLR4-deficient mice was also not influenced by administration of E coli (figure 1F, right). Furthermore, non- and E coli-treated TLR4-deficient mice had comparable frequencies of IL-5-producing CD4 T cells in the airways, whereas E coli administration to wild-type mice again led to significant inhibition of IL-5 production by these cells (figure 1G). We additionally assessed whether NOD1 and NOD2 receptors were involved in recognition of bacterial products and suppression of AAI. Notably neither NOD1- nor Rip2-deficient mice (which lack signalling through both NOD1 and NOD2 receptors) exhibited any differences in the bacterial-mediated suppression of AAI as compared with control mice, excluding a role for these pathways in our model (Supplementary figure 3). Notably, the TLR4-dependent mechanism did not involve TLR desensitisation (Supplementary figure 4). Taken together, our data suggest that E coli induces a TLR4-dependent response in the lung, which leads to suppression of AAI.
Th1 and Treg responses do not play a role in the modulation of AAI by E coli
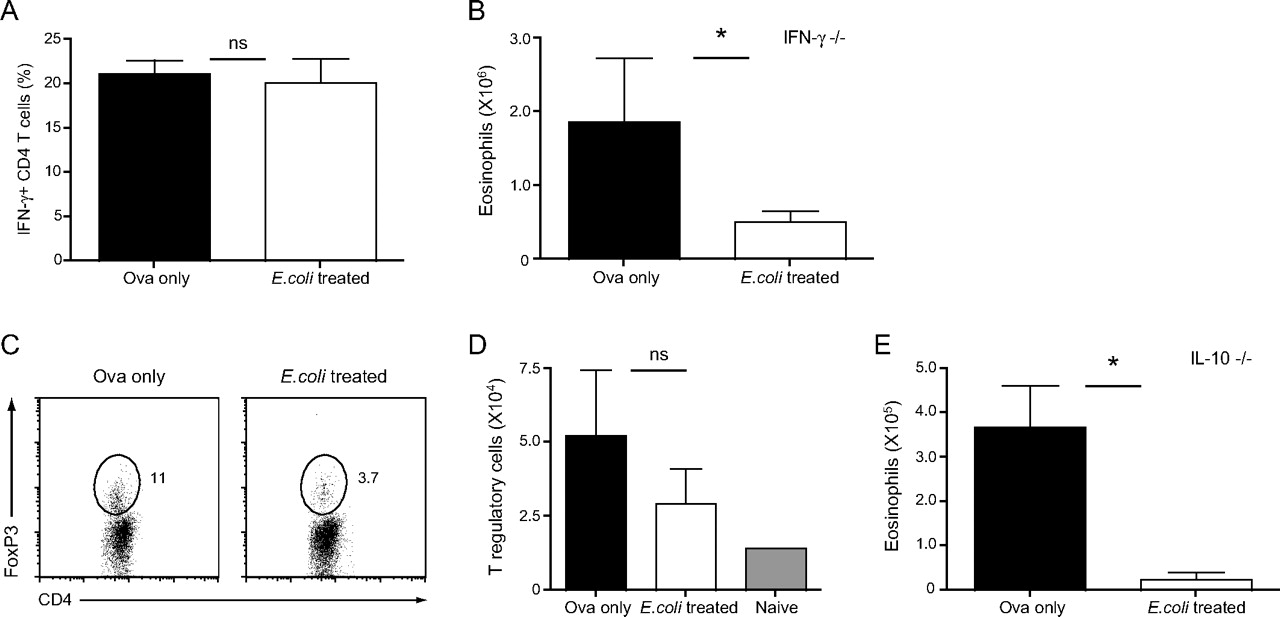
To investigate further the mechanisms underlying the inhibition of AAI by E coli, the role of Th1 responses was assessed. We first measured the proportion of IFNγ-producing CD4 T cells in the airways of non- and E coli-treated mice on day 4 after challenge; however, we did not observe increased frequencies of IFNγ-producing BAL CD4 T cells in mice treated with E coli (figure 2A). Our AAI model was then repeated in IFNγ-deficient mice; E coli treatment could inhibit eosinophil recruitment in mice lacking IFNγ (figure 2B), mimicking the disease outcome observed in wild-type mice (figure 1).

Inhibition of the development of allergic airway inflammation (AAI) is independent of Th1 and regulatory T cell responses. (A) Non-treated and E coli-treated C57BL/6 mice were sensitised and challenged with ovalbumin (OVA). On day 4 after intranasal challenge, bronchoalveolar lavage (BAL) cells were isolated and the proportion of interferon γ (IFNγ)-producing CD4 T cells was determined after restimulation by intracellular staining. IFNγ-deficient mice were treated as described in (A); eosinophil numbers in the BAL were determined (B). (C) Proportion of CD4+FoxP3+ regulatory T cells (Tregs) in the lungs. Values in dot plots represent the percentage of cells detected in the indicated gate. Representative data from one mouse per group are shown. (D) Total Treg numbers in the lung. (E) Eosinophil numbers in BAL of interleukin 10 (IL-10)-deficient mice treated as described in (A). Results are presented as mean±SD. *p <0.05; statistically significant differences. Experiments were repeated 2–3 times with four mice per group.
We next measured Treg frequencies. The proportion of lung FoxP3+CD4 T cells was decreased in E coli-treated mice as compared with control mice (figure 2C). This trend was reflected in the total Treg number (figure 2D), although the difference was not significant. E coli treatment did not increase Treg frequencies in the lung; however, it could still have an influence on their function. We thus analysed the role of the immunoregulatory cytokine IL-10 in our model by treating IL-10-deficient mice with E coli followed by OVA sensitisation. Eosinophil numbers in the BAL of E coli-treated IL-10-deficient mice were drastically reduced when compared with their non-treated counterparts (figure 2E). Our data indicate that suppression of AAI by intranasal E coli administration takes place independently of IFNγ, IL-10 and Tregs.
E coli-induced γδ T cells protect from AHR during AAI
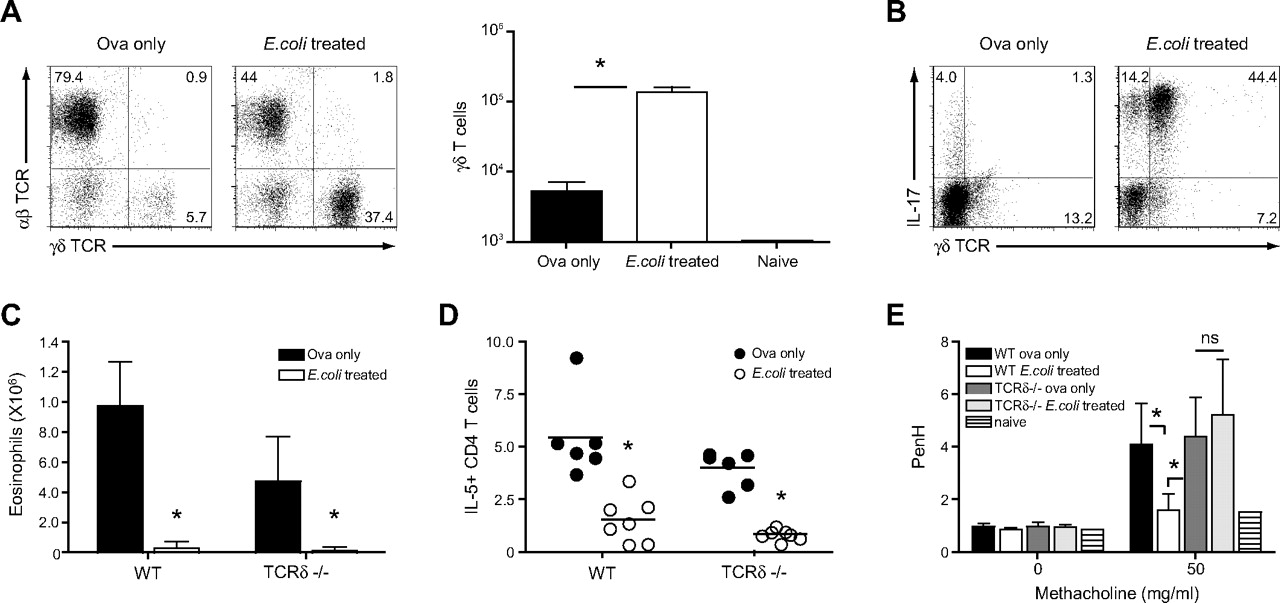
Differential cell counts in the BAL performed on day 4 following challenge with OVA revealed an increased proportion of lymphocytes in E coli-treated mice, and further analysis by flow cytometry identified these cells as γδ T cells (figures 1C and 3A), the majority of which were capable of producing IL-17 (figure 3B).

γδ T cells recruited to the lung upon intranasal challenge with E coli modulate airway hyper-responsiveness (AHR). Non-treated and E coli-treated C57BL/6 mice were sensitised and challenged with ovalbumin (OVA). On day 4 after intranasal challenge with OVA, bronchoalveolar lavage (BAL) cells were isolated. (A) proportion of αβ and γδ T cell receptor (TCR)-bearing T cells as well as total γδ T cell numbers were determined. (B) Cytokine production by γδ T cells was assessed by intracellular staining and flow cytometry. Values in FACS (fluorescence-activated cell sorting) plots represent the percentage of cells detected in each quadrant. Representative data from one mouse per group are shown. (C) C57BL/6 and TCRδ-deficient mice were treated as described above, and eosinophil numbers in the BAL were determined 4 days after OVA challenge. (D) Additionally, the proportion of interleukin 5 (IL-5)-producing CD4 T cells in the BAL was assessed by intracellular staining and flow cytometry. (E) On day 3 after intranasal challenge with OVA, non-treated and E coli-treated C57BL/6 and TCRδ mice were exposed to increasing doses of methacholine, and AHR was determined in a full body unrestrained plethysmograph. Results represent values from single animals or are presented as mean±SD. *p <0.05; statistically significant differences. Experiments were repeated 2–3 times with 4–8 mice per group. WT, wild type.
We next investigated whether the presence of IL-17-secreting γδ T cells was playing a role in mediating the suppression of the AAI. Accordingly, E coli-treated wild-type as well as TCR-δ-deficient mice were immunised and challenged with OVA and compared with their non-treated counterparts. Suppression of eosinophil recruitment and Th2 cytokine production was independent of the presence of γδ T cells (figure 3C, D). E coli-treated wild-type mice displayed decreased AHR as compared with control mice; however, in the absence of γδ T cells, both groups of mice developed AHR in response to the bronchoconstrictor methacholine (figure 3E). These data indicate that while γδ T cells do not impact cell recruitment in this model, they are capable of specifically influencing AHR.
E coli-treated mice display normal DC migration and CD4 T cell priming in lung-draining LNs
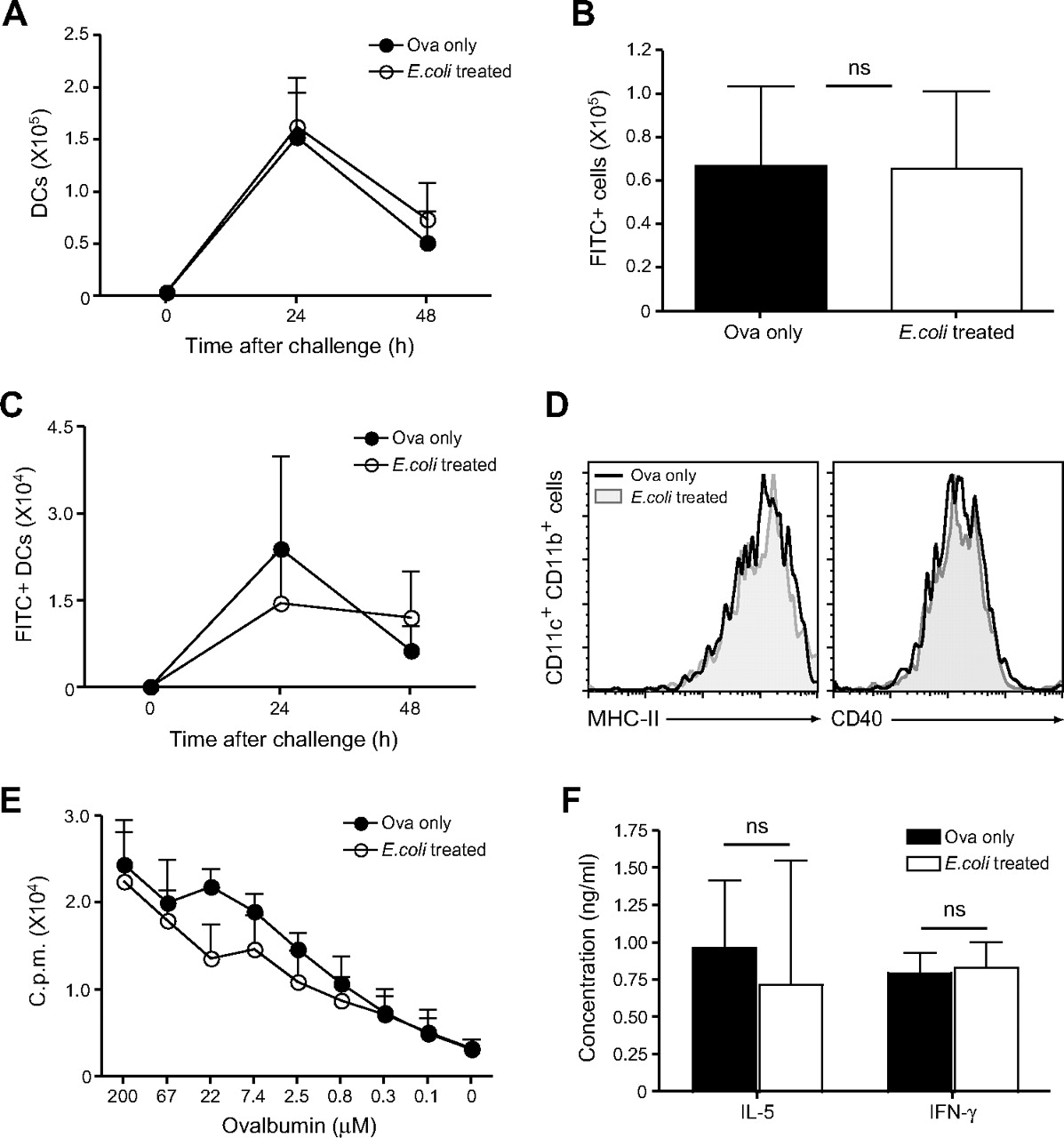
DCs take up antigen in the airways and rapidly transport it to the lung-draining LNs where presentation to specific T cells can occur; a normal DC migratory capacity is crucial for the induction of immune responses of the respiratory tract.19 The influence of E coli administration on DC migration and antigen transport to the draining LNs in the course of an asthmatic response was evaluated. First, total DC numbers were determined in the mediastinal LNs of non- and E coli-treated mice 24 and 48 h after OVA challenge. Migrating DCs were identified according to the expression of high levels of CD11c and CD11b.2 20 Total DC numbers were comparable between non-treated asthmatic mice and mice treated with E coli prior to asthma induction (figure 4A).

Dendritic cell (DC) migration to the draining lymph node and CD4 T cell priming are not impaired by administration of bacteria. Non-treated and E coli-treated C57BL/6 mice were challenged intranasally with fluorescein isothiocyanate (FITC)-labelled ovalbumin (OVA). After 24 and 48 h, single cell suspensions of lung-draining lymph nodes (LNs) were prepared. (A) Total DC numbers in the draining LNs were determined according to CD11c and CD11b expression. (B) Total FITC+ LN cell numbers at 24 h after challenge. (C) Total FITC+ DC numbers were determined as described in (A). (D) Major histocompatibility complex (MHC) class II and CD40 expression on LN CD11c+CD11b+ DCs 24 h after challenge. (E) On day 4 after intranasal challenge with OVA, CD4 T cells were purified from the draining LNs and cultured with splenic DCs and different concentrations of OVA protein. Proliferation was determined by [3H]thymidine incorporation. Alternatively, interleukin 5 (IL-5) and interferon γ (IFNγ) secretion in the culture supernatant was determined by ELISA (F). Results are presented as mean±SD. Experiments were repeated 2–3 times with four mice per group.
Next, antigen transport to the draining LNs was analysed by challenging the mice with FITC-conjugated OVA. Antigen-bearing FITC+ cells were detected in the mediastinal LNs 24 h after challenge, and again comparable cell numbers could be observed between PBS- and E coli-treated mice (figure 4B). Moreover, no significant impairment in antigen uptake and transport by CD11chighCD11bhigh DCs was observed in E coli-treated mice (figure 4C), and expression of activation markers such as major histocompatibility complex (MHC) class II and CD40 was also normal when compared with non-treated asthmatic mice (figure 4D). Additionally, we evaluated the proliferative capacity of CD4 T cells in the draining LNs of non-treated and E coli-treated mice. CD4 T cells were isolated and cultured with freshly isolated splenic DCs and different concentrations of OVA; proliferation was assessed after 3 days by [3H]thymidine incorporation. As shown in figure 4E, CD4 T cells isolated from E coli-treated mice were able to proliferate to a similar extent compared with T cells from non-treated asthmatic mice. As an additional measurement of T cell effector function, IL-5 and IFNγ production by LN CD4 T cells was measured in the supernatant of the 3 day culture with DCs and 200 μM OVA. Cytokine production by CD4 T cells was unaffected in E coli-treated mice as compared with non-treated, allergic mice (figure 4F). Taken together our results suggest that administration of the bacteria does not lead to suppression of airway inflammation via the inhibition of DC maturation and T cell priming in the draining LNs.
Intranasal E coli administration alters DC function in the lung
In the lung, stimulation of effector T cells by local DCs is crucial for the inflammatory response.3 21 We hypothesised that E coli administration could negatively influence DC function in the lung tissue as opposed to the draining LNs, where antigen uptake and T cell priming were normal following exposure to the bacteria. Antigen uptake by antigen-presenting cells (APCs) in the lung was analysed by challenging control non-treated as well as E coli-treated mice with OVA–FITC. Lungs were isolated 24 h after challenge and FITC+ cells were analysed by flow cytometry; total FITC+ cell numbers were comparable between E coli- and non-treated mice (figure 5A). We further characterised FITC+ cells according to their expression of the surface molecules CD11b and CD11c, which are expressed at different levels by APCs in the lung.2 In non-treated mice the largest proportion of antigen-loaded cells consisted of CD11c+CD11b+ cells (figure 5B), which correspond to DCs with high antigen uptake and presentation capacity.2 In contrast, in the lung of E coli-treated mice, only a small fraction of DCs was able to take up the antigen, which was instead mostly present within autofluorescent, CD11c+CD11binter resident macrophages.22 We further confirmed this result by analysing total OVA–FITC+ cell numbers; E coli-treated mice had significantly decreased numbers of FITC+CD11c+CD11b+ DCs when compared with their non-treated, allergic counterparts (figure 5C, left). Conversely, a higher number of antigen-loaded CD11c+CD11binter cells could be observed after bacterial administration (figure 5C, right).

Lung dendritic cells (DCs) display impaired maturation and effector function following inhalation of E coli. Mice were sensitised with ovalbumin (OVA) and challenged intranasally with fluorescein isothiocyanate (FITC)-labelled OVA, and lung cells were isolated after 24 h. (A) Total FITC+ lung cells. (B) Uptake of OVA–FITC by different CD11b- and CD11c-expressing lung cell populations. Values in FACS (fluorescence-activated cell sorting) plots represent the percentage of cells detected in the indicated gates. Representative data from one mouse per group are shown. (C) Total numbers of FITC+ cells of the indicated populations. (D) Expression of major histocompatibility complex (MHC) class II was assessed by flow cytometry. (E) CD11c+CD11b+ and CD11c+CD11binter cells were FACS sorted from the lung 24 h after intranasal OVA challenge, and cultured in vitro with OVA-specific CD4 T cells isolated from transgenic OT-II mice. Proliferation was assessed by [3H]thymidine incorporation (left). Alternatively, CD4 T cells were restimulated with phorbol myristate acetate PMA/ionomycin, and interferon γ (IFNγ) production was determined by intracellular staining (right). (F) Total lung CD11c+CD11b+ numbers. (G) Non-treated and E coli-treated mice were sensitised with OVA and challenged intranasally with OVA-pulsed or unpulsed bone marrow-derived DCs 7 days later. On day 4 after DC administration, bronchoalveolar lavage (BAL) was analysed for eosinophil infiltration as well as interleukin 4 (IL-4) and IL-5 production by CD4 T cells. (H) A 2 μg of tumour necrosis factor α (TNFα) was administered intranasally to E coli-treated mice concomitantly with the challenge with OVA. Eosinophil influx in the BAL was determined on day 4 after challenge. Results represent values from single animals or are presented as mean±SD. *p < 0.05; statistically significant differences. Experiments were repeated 2–3 times with 4–8 mice per group.
We next analysed the expression of activation markers on the surface of lung DCs as well as CD11c+CD11binter cells following OVA challenge. In line with previous reports, CD11c+CD11b+ DCs of non-treated, allergic mice displayed high surface expression levels of MHC class II; however, DCs isolated from E coli-treated mice expressed significantly lower levels of this activation marker (figure 5D, left panel). Of note, no difference in CD86 and CD40 expression by DCs was observed between E coli-treated and allergic mice (data not shown). In contrast to DCs, CD11c+ macrophages only expressed intermediate levels of MHC class II, and no difference could be observed between the two groups (figure 5D, right panel).
The capacity of the different lung APCs in presenting antigen to naïve OVA-specific T cells was analysed in vitro. Twenty-four hours after OVA challenge, CD11c+CD11b+ and CD11c+CD11binter cells were sorted from lung cells of non- and E coli-treated mice followed by culture in equal numbers with CD4 T cells isolated from transgenic OT-II mice. Three days later, proliferation of OVA-specific cells and cytokine production were evaluated. Proliferation of CD4 T cells cultured with CD11c+CD11b+ DCs from E coli-treated mice (E coli-DCs) was drastically reduced when compared with the non-treated allergic controls (figure 5E, left). CD11c+CD11binter cells isolated from non-treated mice were less capable of inducing T cell expansion than CD11c+CD11b+ DCs, and this population was also negatively regulated by treatment with the bacteria. Moreover, lower proportions of CD4 T cells were producing IFNγ when stimulated by E coli-DCs compared with control DCs, whereas CD11c+CD11binter cells from both non-treated and E coli-treated mice poorly induced IFNγ production (figure 5E, right).
The influence of E coli administration on DC migration to the lung following antigen inhalation was also analysed. Notably we observed a rapid increase in CD11c+CD11b+ DC numbers in the lung 24 h after challenge with OVA, yet no significant difference between non- and E coli-treated mice could be detected (figure 5F).
Finally, we attempted to overcome the suppression of airway inflammation by delivering fully matured, antigen-presenting DCs directly to the lung at the time of challenge. In vitro generated BM-DCs, which closely resemble CD11c+CD11b+ lung DCs, were pulsed with OVA and administered intranasally to non-treated as well as E coli-treated mice previously sensitised with OVA in alum; control mice were challenged with unpulsed DCs. E coli-treated mice displayed decreased eosinophil infiltration as well as Th2 cytokine production even following the delivery of OVA-pulsed DCs as compared with their non-treated counterparts (figure 5G). Furthermore, intranasal delivery of tumour necrosis fatcor α (TNFα) together with the OVA challenge to induce in situ DC activation23 did not restore the allergic airway inflammation in E coli-treated mice (figure 5H).
Cytokine production by effector Th2 cells in the lung is impaired in E coli-treated mice
We sought to analyse whether E coli treatment could affect migration of antigen-specific effector Th2 cells to the site of inflammation and in situ cytokine production. OVA-specific CD4 T cells from OT-II transgenic mice (expressing the leucocyte marker CD45.2+) were differentiated in vitro to Th2 effector cells, and subsequently injected intravenously into mice previously treated with E coli or non-treated mice. Mice were then challenged intranasally with OVA, and the recruitment of transferred cells to the airways was determined. The number of transferred T cells in BAL, lung and draining LNs of E coli-treated mice was not decreased (figure 6A). Despite similar or even increased CD45.2+ Th2 cell numbers in the airways, mice treated with the bacteria displayed significantly lower eosinophil frequencies in the BAL, indicating that prior E coli treatment was able to impair the effector function of polarised Th2 cells in the lung (figure 6B). We observed reduced frequencies of IL-4- and IL-5-producing CD45.2+ T cells in the lung of E coli-treated mice as compared with non-treated mice challenged with OVA (figure 6C). IL-13 and IL-5 could be detected in the airways of non-treated, OVA-challenged mice, but not in E coli-treated or non-challenged control mice (figure 6D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
T helper 2 (Th2) cells recruited to the lung of E coli-treated mice display impaired cytokine production. CD45.2+CD4 T cells isolated from the spleen of ovalbumin (OVA)-transgenic OT-II mice were polarised to Th2 cells in vitro and adoptively transferred into CD45.1+C57BL/6 mice previously treated with E coli or into non-treated mice. On the same day, and for the following 3 days, adoptively transferred mice were challenged intranasally with ovalbumin (OVA); control mice did not receive OVA (grey bar). At 24 h after the last challenge, bronchoalveolar lavage (BAL), lung and draining lymph nodes (LNs) were isolated. (A) Total CD45.2+ T cell numbers were determined in the indicated tissue compartments. (B) Eosinophil proportion in the BAL. (C) Cytokine production by transferred lung CD45.2+ cells after restimulation. Representative data from one mouse per group are shown. (D) Interleukin 13 (IL-13) and IL-5 secretion in BAL fluid was determined by ELISA. Results represent values from three different mice per group and are presented as mean±SD. *p <0.05; statistically significant differences. Experiment was repeated twice with three mice per group.
Discussion
Development of an asthmatic response involves antigen uptake by airway DCs and their migration to the draining LNs. In the lymphoid tissue, DCs present the antigen to specific naïve and memory T cells, leading to the generation of effector Th2 cells, which then migrate to the site of inflammation.24 In the inflamed lung, Th2 cells require further activation by DCs to produce inflammatory cytokines efficiently.3 25 In the current study, we have demonstrated that irrespective of whether mice have been exposed to E coli, DCs readily transported antigen to the draining LNs and exhibited normal maturation. However, we observed a dramatic reduction of Th2 cytokine production by CD4 T cells in the BAL and lung of E coli-treated mice, suggesting that activation of these cells was being subverted locally. We speculated that DC function in the lung as opposed to the LNs was affected. Indeed, following intranasal challenge with FITC-labelled OVA, CD11c+CD11b+ DCs from the lung of E coli-treated mice took up significantly less antigen when compared with non-treated, allergic mice. Moreover, although total DC numbers were comparable between non-treated and E coli-treated mice, the same DCs expressed lower levels of MHC class II molecules, indicating that bacterial administration influenced the maturation of DCs and their presentation capacity, without affecting their survival or recruitment to the inflamed lung. We propose that alteration of lung DC maturation and effector function might be a crucial mechanism for the inhibition of effector CD4 T cell activation and, consequently, AAI following intranasal administration of bacteria.
Interestingly, most of the antigen administered to the airways of E coli-treated mice was taken up by resident macrophages. This lung APC population was previously shown to be less efficient than CD11c+CD11b+ DCs in activating naïve T cells in vitro,22 and our data are in line with these findings. We conclude that upon administration of the bacteria, antigen presentation by CD11c+CD11b+ DCs with impaired local functionality, in addition to poor presentation by CD11c+ macrophages, fails to effectively activate T cells that are recruited to the lung. The transfer of cytokine-producing OVA-specific Th2 cells suggested that even though the recruitment was not decreased, in vivo cytokine production by these Th2 cells was significantly impaired in E coli-treated mice.
Considering the timing of bacterial administration and asthma induction in our model, the limited life span of lung CD11c+CD11b+ DCs22 and the constant recruitment of new DCs from the blood to the airways during inflammation,26 we consider a direct regulation of DC function by E coli unlikely to be the only effect in mediating the suppression of inflammation. We attempted to reconstitute lung DC function and thus the induction of AAI in E coli-treated mice by challenging mice with fully competent, OVA-pulsed BM-DCs instead of OVA protein alone. In line with previous reports, OVA-pulsed BM-DC administration induced AAI in sensitised mice,3 but failed to do so in mice previously treated with the bacteria. These results suggest that the lung environment exhibits a suppressive effect upon DCs that might remain constant following E coli administration or become reactivated at the time of antigen challenge. Such a phenomenon would further explain the long-lasting suppression of AAI following treatment with the bacteria. Notably, in contrast to DCs, the turnover of pulmonary macrophages can be several weeks or even months,22 further indicating that these cells might play a role in AAI inhibition over longer time periods.
Intact TLR4 signalling was a crucial factor for the inhibition of asthma in our model; TLR4-deficient mice treated with the bacteria displayed the same extent of airway inflammation as non-treated, OVA-sensitised mice. These results indicate that LPS-dependent innate immune responses triggered by E coli in the lung are essential for modulating the allergic response. Of note, a role for endotoxin in the regulation of allergic responses has been postulated, although, depending on the dose and timing of the exposure, the opposite effect could also be observed.5 18 27 Interestingly, inhibition of asthma development via environmental exposure to endotoxin was associated with increased Th1 cell-mediated responses,28 and the same result was observed when mice were treated intranasally with high doses of LPS alone.23 Moreover, Kuipers et al showed that immunisation with OVA-pulsed DCs previously treated with LPS could induce higher levels of IFNγ and eventually dampen airway Th2 responses following intranasal challenge with OVA.29 However, in our model, E coli administration did not induce a shift to a Th1 immune response, and suppression of allergic inflammation was independent of IFNγ.
Taken together, our data suggest there is a multicomponent mechanism underlying the inhibition of AAI following intranasal administration of non-pathogenic bacteria such as E coli. This process does not depend on a switch to Th1 responses, or on the induction of elevated regulatory T cell frequencies and IL-10 production. Rather, the recruitment of γδ T cells appears to play a role in controlling AHR, and a TLR4-dependent mechanism induces a suppressive lung environment in which DC function is altered, and consequently the induction of Th2 effector function locally in the lung is abrogated.
Acknowledgments
We would like to thank Cornelius Fischer for FACS sorting.
References
Supplementary materials
Web Only Data thx.2010.152512
Files in this Data Supplement:
Footnotes
See Editorial, p 744
Linked articles 160853.
Funding This work has been funded by the Swiss National Science Foundation (SNF) 310000-116675 and ETH 0-08-2, 0-20400-07 research grants. BJM is supported by a Cloëtta Medical Research Fellowship.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.