Article Text

Abstract
Background The current lifetable approach to survival estimation is favoured by CF registries. Recognising the limitation of this approach, we examined the utility of a parametric survival model to project birth cohort survival estimates beyond the follow-up period, where short duration of follow-up meant median survival estimates were indeterminable.
Methods Parametric models were fitted to observed survivorship data from the US CF Foundation (CFF) Patient Registry 1980–1994 birth cohort. Model-predicted median survival was estimated. The best fitting model was applied to a Cystic Fibrosis Registry of Ireland dataset to allow an evaluation of the model's ability to estimate predicted median survival. This involved a comparison of birth cohort lifetable predicted and observed (Kaplan–Meier) median survival estimates.
Results A Weibull model with main effects of gender and birth cohort was developed using a US CFF dataset (n=13 115) for which median survival was not directly estimable. Birth cohort lifetable predicted median survival for male and female patients born between 1985 and 1994 and surviving their first birthday was 50.9 and 42.4 years respectively. To evaluate the accuracy of a Weibull model in predicting median survival, a model was developed for the 1980–1984 Cystic Fibrosis Registry of Ireland birth cohort (n=243), which had an observed (Kaplan–Meier) median survival of 27.7 years. Model-predicted median survival estimates were calculated using data censored at different follow-up periods. The estimates converged to the true value as length of follow-up increased.
Conclusions Accurate prognostic information that is clinically critical for care of patients affected by rare, life-limiting disorders can be provided by parametric survival models. Problems associated with short duration of follow-up for recent birth cohorts can be overcome using this approach, providing better opportunities to monitor survival and plan services locally.
- Cystic fibrosis
- expectation of life
- mortality
- parametric model
- survival analysis
- clinical epidemiology
- tuberculosis
- bacterial infection
- bronchiectasis
- clinical epidemiology
- respiratory muscles
- short burst oxygen therapy
- thoracic surgery
- bronchiectasis
- health economist
- COPD mechanisms
- COPD exacerbations
- lung physiology
- pulmonary rehabilitation
- respiratory measurement
- respiratory muscles
- asthma
- asthma mechanisms
- cough/mechanisms/pharmacology, infection control
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Cystic fibrosis
- expectation of life
- mortality
- parametric model
- survival analysis
- clinical epidemiology
- tuberculosis
- bacterial infection
- bronchiectasis
- clinical epidemiology
- respiratory muscles
- short burst oxygen therapy
- thoracic surgery
- bronchiectasis
- health economist
- COPD mechanisms
- COPD exacerbations
- lung physiology
- pulmonary rehabilitation
- respiratory measurement
- respiratory muscles
- asthma
- asthma mechanisms
- cough/mechanisms/pharmacology, infection control
Key messages
What is the key question?
Can median survival estimates be projected beyond observed survivorship data for recent birth cohorts of people with cystic fibrosis (CF) when more than half of the cohort is still alive?
What is the bottom line?
Parametric modelling of CF birth cohort data can overcome the challenge of estimating birth cohort median survival following a short follow-up period.
Why read on?
Accurate prognostic information can be provided by parametric models of CF birth cohort data, providing better opportunities to monitor and plan CF services.
Introduction
Cystic fibrosis (CF) registries are the principal source of data for analysis of CF survival.1 The US CF Registry is one of the largest and longest established, reporting data from over 25 000 patients receiving care at CF Foundation (CFF) accredited centres since 1982. Median survival is a common measure used by registries to monitor life expectancy for patients with CF. Median survival estimation follows an entire CF population until half are deceased, and is the age at which half of the population have died. Median survival can be derived using two different approaches: current (period) and cohort lifetables.2–8 However, the application of each has limitations in CF9 and has resulted in failure to adopt a simple standardised approach internationally.
The current lifetable approach takes age-specific mortality rates observed in a calendar year and estimates a ‘current lifetable predicted median survival’ for a hypothetical population by assuming those mortality rates validly estimate future rates and remain constant over the population's lifetime.10 However mortality rates are not static; notable improvements have been observed during childhood in recent years.9 11
The alternative method uses a birth cohort lifetable to estimate ‘birth cohort lifetable observed median survival’ and requires all CF births in a given period to be identified and followed until death or until more than 50% are deceased for estimation of median survival. Cohorts of recent births will not have an estimate for many years because more than half are still alive.12
Because of the inability of many CF registries to observe birth cohort lifetable median survival for cohorts of recent births, the current lifetable approach is more frequently applied to monitor temporal trends in median survival.3 4 For instance, in 2008, the US CFF Patient Registry reported that 46.3% of patients with CF were aged ≥18 years,3 and that the current lifetable predicted median survival estimate was 37·4 years.
We examined the utility of a parametric survival model to obtain estimates of birth cohort lifetable predicted median survival beyond the follow-up period, where short duration of follow-up by CF registries meant birth cohort lifetable observed median survival estimates were otherwise indeterminable.
Study data
This analysis is based on CF data from two patient registries. The first dataset was derived from the US CFF Patient Registry. This registry confirms deaths and death dates by direct contact with the CF centres. US death certification records were unavailable and patients lost to follow-up in 2007 (did not attend a CFF clinic in that year) were therefore excluded from this analysis. The second dataset was defined using data from the Cystic Fibrosis Registry of Ireland (CFRI) and a listing of deaths compiled from three sources: Central Statistics Office registered deaths when CF was reported as the underlying cause (1980–2007), CF centre records of CF deaths (2002–2007), and CF patient association recorded deaths (1986–2007). Study patients not reported as deceased were therefore presumed to be alive.
Each study population comprised a birth cohort of patients with CF born between 1980 and 1994 and diagnosed by 31 December 2007 (‘study end date’). Patients who underwent solid-organ transplantation were not excluded or censored at transplantation to ensure consistency across datasets. The following data were provided for each patient: year of birth, gender, vital status on study end date, and survival time from birth to death or study end date. We calculated survival for all study patients from date of birth, although left truncation at either date of diagnosis or entry into the CF registry would be the most appropriate treatment of the data. Left truncation was precluded by the absence of date of diagnosis data for Ireland decedents not enrolled on the CFRI (ie, study patients identified from registered death records). Although the US and Irish CF registries report the highest CF population ascertainment rates internationally (approximately 90%), median survival estimates may be unavoidably biased because all people with CF born in a specific time period may not have been detected and followed over the period.13
Table 1 shows differences between CFF and CFRI birth cohorts. The number of US registry-enrolled patients with CF born between 1980 and 1994 is 20 times that of the Irish registry, and the number of birth cohort deaths by 2007 is nearly twice as many for CFRI (30.2%) compared with CFF (17.4%).
Characteristics of patients born between 1980 and 1994 enrolled on the US Cystic Fibrosis Foundation Patient Registry and the Cystic Fibrosis Registry of Ireland, with known vital status, and who were diagnosed with cystic fibrosis by 2007
Deaths in CFF and CFRI birth cohorts were then examined using three, 5-year birth cohorts (1980–1984, 1985–1989, 1990–1994) (table 2). Such a small proportion of deaths occurred during the follow-up period (13 years) of the 1990–1994 birth cohort that it was deemed appropriate to combine the 1985–1989 and 1990–1994 birth cohorts. Subsequently, birth cohort lifetables were calculated and survivorship curves generated for each country, for one 5-year and one 10-year birth cohort (1980–1984, 1985–1994) using the Kaplan–Meier method (SURV and SURVFIT functions) in R software version 2·10·1 (downloadable from http://www.r-project.org/).14 Although less than 50% of the CFRI 1980–1984 birth cohort had died by 2007, the observed median survival could be determined using the Kaplan–Meier method. The point in time to which 50% of the cohort survives is not necessarily the same as the median for the Kaplan–Meier derived cumulative survival function. This can occur when a lifetable contains censored observations prior to the 50th percentile of the cumulative survival function.
Deaths observed by 2007 for three 5-year birth cohorts (1980–1984, 1985–1989, 1990–1994) of patients enrolled on the US Cystic Fibrosis Foundation Patient Registry and the Cystic Fibrosis Registry of Ireland
Analysis strategy
Various parametric models were used to fit survival curves for birth cohort lifetable predicted median survival estimation. Because of its large size, the pooled US CFF birth cohort dataset (1980–1994) was used to select the best fitting parametric survival model. Model parameters were then estimated for the US dataset using the variables gender and birth cohort (1980–1984, 1985–1994). The model structure that best fit the US dataset was applied to the CFRI dataset in order to evaluate the ability of the model to estimate birth cohort lifetable predicted median survival. Model parameters were calculated independently for the CFRI dataset.
Introduction to model development
A parametric model assumes that observed data follows a particular distribution (eg, exponential, Weibull) and that one of those different distributions will best fit CF survival data. Unlike non-parametric models (eg, Cox proportional hazards), extrapolation of survival estimates beyond the study observation period is possible using a parametric approach.15 However, the benefit of projecting a survival estimate beyond observed data is tempered by the sensitivity of the estimate (and its associated degrees of uncertainty) to the underlying model structure and is governed by the size and duration of follow-up of the observed birth cohort.
Patients with CF who died before their first birthday were excluded during model development because high mortality rates during the first year of life led to poor model fit. Additionally, this action unintentionally benefitted the analysis because potential bias introduced by the unavoidable omission of unrecognised infant deaths from CFF3 16 and CFRI datasets was avoided. The parametric model and the corresponding birth cohort lifetable predicted median survival estimates are for patients with CF surviving their first birthday.
Model parameter estimation, which was performed independently for CFF and CFRI datasets, involved adding gender and birth cohort (1980–1984, 1985–1994) as potential explanatory variables to the model. Differences between the two cohorts and between male and female survival estimates were examined (table 3). Fully separated models (stratified by both gender and birth cohort) were excluded from the analysis a priori because of the small proportion of deaths observed in US male patients born between 1985 and 1994 (9%) and US female patients born between 1985 and 1994 (12%). Parametric model fitting and parameter estimation was performed using the SURVREG function in R.
Akaike Information Criteria (AIC) for Weibull models with explanatory variables of gender and birth cohort (1980–1984 and 1985–1994) for patients enrolled on the US Cystic Fibrosis Foundation Patient Registry surviving their first birthday
Model selection and fitting using US CF Foundation Patient Registry data
Four model structures, each with a distinct underlying survivorship function for fitting observed data to parametric models, were deemed suitable for examination: exponential, Weibull, log-logistic and log-normal. Initially, all US CFF data were entered into the model, pooling observations across birth cohorts (1980–1994) and gender to help decide which model structure was most appropriate for the data (table 3, model A).
Akaike's Information Criterion (AIC) was used to determine which underlying model best fitted observed survivorship, with lower values indicating a better fit. Because log-likelihood is an additive property of a model and dataset,17 AIC values can be summed across non-overlapping stratifications of the same dataset to yield an overall AIC value for the entire dataset.18 Birth cohort lifetable predicted median survival and 95% CIs were estimated using the SURVREG function reverse prediction method.14
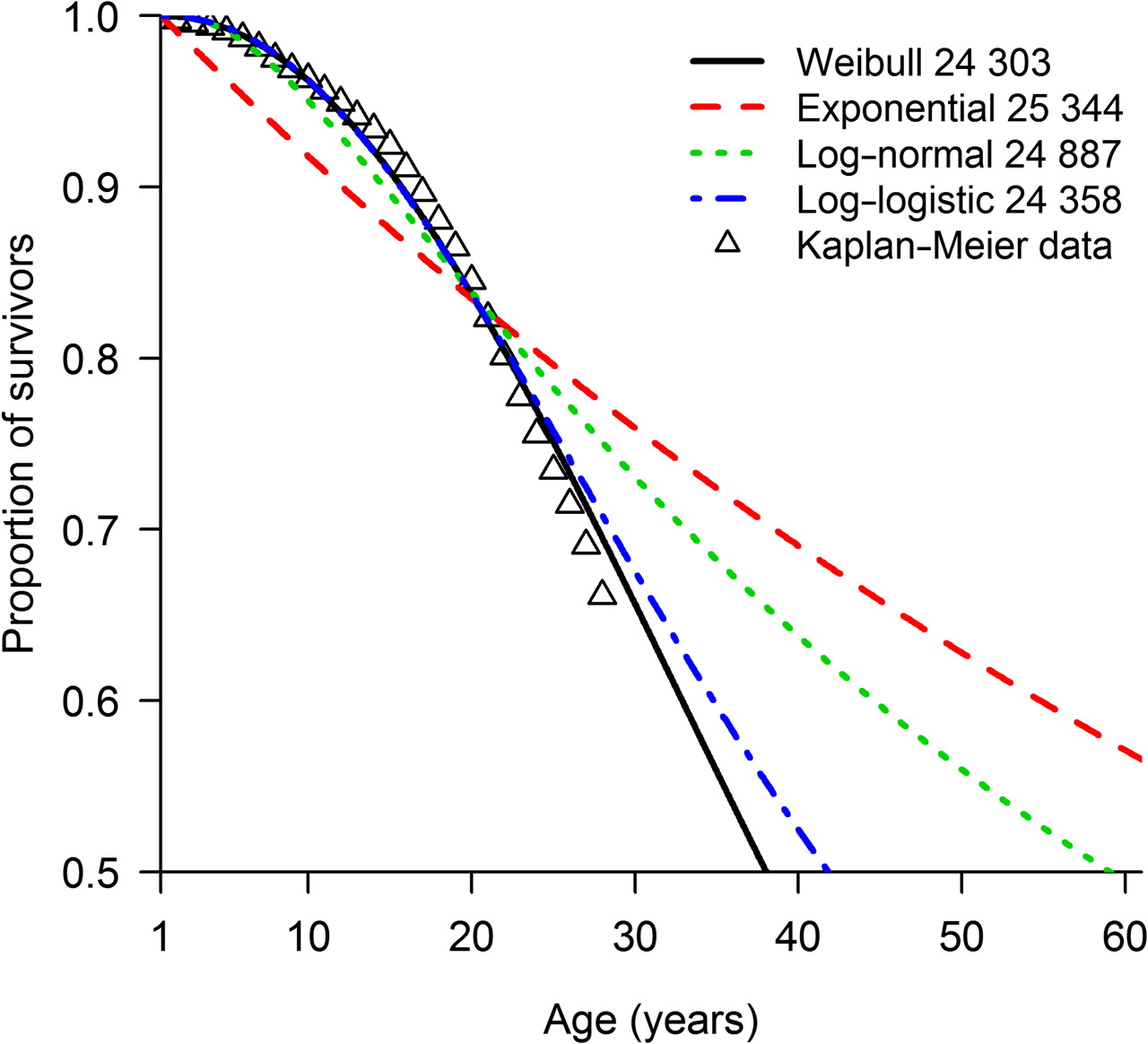
Using the pooled US dataset (1980–1994) of patients with CF surviving their first birthday (13 115 observations), the Weibull (AIC =24 060.3) model best explained the data (figure 1, table 3). On examining differences in survivorship between gender and birth cohorts, the best model contained gender and birth cohort (1980–1984, 1985–1994) as the main effects under a Weibull distribution (table 3, model D), and was marginally better than the model containing gender and birth cohort with an interaction effect (model E).

Kaplan–Meier survivorship curve and parametric models with associated Akaike Information Criteria for patients born between 1980 and 1994 enrolled on the US Cystic Fibrosis Foundation Patient Registry who survived their first birthday.
Under the model with main effects of gender and birth cohort, US male patients were shown to live longer than female patients and survival improved between the 1980–1984 and 1985–1994 birth cohorts (figure 2A, table 4). Birth cohort lifetable predicted median survival for patients surviving their first birthday was 37.8 and 31.5 years for male and female patients born between 1980 and 1984, and 50.9 years and 42.4 years for male and female patients born between 1985 and 1994.

Kaplan–Meier survivorship curves (females = ○, males = ∆) with fitted Weibull model with main effects of gender (female = black line, male = red line) and birth cohort and corresponding 95% CIs (dashed lines) for patients with cystic fibrosis surviving their first birthday and enrolled on the US Cystic Fibrosis Foundation Patient Registry (A) and the Cystic Fibrosis Registry of Ireland (B).
Birth cohort lifetable predicted median survival estimates and 95% CIs for a Weibull model with main effects of gender and birth cohort for patients enrolled on the US Cystic Fibrosis Foundation Patient Registry surviving their first birthday
Parameter fitting using CFRI data
Because the Weibull model was deemed to best fit US CFF survivorship data, the model was applied to the CFRI dataset. Estimation of CFRI model parameters took place independently of the CFF model parameter estimation. A dataset of 624 CFRI patients with CF born between 1980 and 1994 and surviving their first birthday was entered into the model. An AIC of 1659 was determined for the null Weibull model (ie, no explanatory factors were used in the model). Inclusion of explanatory factors improved model fit and a Weibull model containing gender and birth cohort as the main effects was the best fitting (AIC=1633).
Birth cohort lifetable predicted median survival estimates for male and female patients surviving their first birthday were 32.3 and 24.7 years respectively for those born between 1980 and 1984, and 51.1 and 39.0 years for those born between 1985 and 1994 (figure 2B, table 5). Predicted median survival estimates increased between the birth cohorts by 18.8 years for male patients and 14.3 years for female patients.
Birth cohort lifetable predicted median survival estimates and 95% CIs for a Weibull model with main effects of gender and birth cohort for patients enrolled on the Cystic Fibrosis Registry of Ireland surviving their first birthday
Comparison of CFRI birth cohort lifetable observed (Kaplan–Meier) and predicted median survival estimates
While birth cohort lifetable predicted median survival estimates were calculated for both CFF and CFRI study populations, the latter dataset allowed an evaluation of the ability of the parametric model to predict birth cohort lifetable observed median survival. A birth cohort lifetable observed median survival estimate was derived for CFRI patients with CF born between 1980 and 1984 and surviving their first birthday using the Kaplan–Meier method (see Study data). Model-estimated birth cohort lifetable predicted median survival was compared with the actual observed (Kaplan–Meier) median survival to determine whether the selected model was capable of reliably estimating predicted median survival. A full evaluation of the suitability of the model for estimating survival will only be possible once a dataset becomes available which has followed all patients in a birth cohort until death.
As the actual (Kaplan–Meier) median survival of the CFRI 1980–1984 birth cohort was observed for male patients, but not female patients, the comparison of model-predicted and observed median survival estimates required male and female birth cohort datasets to be pooled. A Weibull model was applied to the CFRI 1980–1984 birth cohort consisting of 243 patients with CF surviving their first birthday (cf, table 3, model G: 1980–1984). The performance of the model in predicting median survival after short duration of follow-up was examined by censoring observed survivorship data (figure 3). Initially, survivorship data were censored after 5 years; therefore only follow-up data to the 31 December 1989 were used in the model. A birth cohort lifetable predicted median survival of 40.0 years was estimated based on only 5 years of follow-up.

{kind=link}
{kind=link}
{kind=link}
Median predicted survival and 95% CIs for patients born between 1980 and 1984 enrolled on the Cystic Fibrosis Registry of Ireland and surviving their first birthday, estimated under a Weibull model during a 23-year follow-up period, in which follow-up was censored in each calendar year. The actual (Kaplan–Meier) median survival for patients with cystic fibrosis surviving their first birthday observed at the study end point (2007) is also shown.
The follow-up period was then extended a year at a time until 2007. A new model was developed each time and model-predicted median survival was re-estimated on each occasion. Convergence of the birth cohort lifetable predicted to the observed median survival estimate (for patients with CF surviving their first birthday) of 27.7 years (lower 95% CI: 24.8, upper 95% CI: indeterminate) was good, particularly from 1995 onwards, which corresponds to 20% observed mortality in the 1980–1984 birth cohort.
Discussion
By developing parametric models of CF birth cohort data to project survivorship, we have started to overcome the challenge of median survival estimation associated with short duration of follow-up of recent cohorts. This approach is suited to survival estimation for disorders where better treatment has resulted in continued improvement in survivorship. Although limitations remain when estimating survival in rare disease populations, regardless of methodological approach,9 we have shown that our model can be applied to registry datasets of variable size, demonstrating its utility as a tool to estimate survival at a local level.
Better treatment has resulted in continued improvement in CF survival, particularly in childhood, yet the current lifetable method, which applies static age-specific mortality rates to a hypothetical population in order to project estimates of life expectancy, is favoured by CF registries. At best, annual median predicted CF survival estimates derived by current lifetables can only provide a snapshot of a moving target.13 For conditions with fluctuating age-specific mortality rates, longitudinal techniques, that is, birth cohort lifetables, may be more appropriate for monitoring survival. However, prospective follow-up of birth cohorts by many CF registries has not yet yielded an observed median survival. Birth cohort follow-up can be undertaken retrospectively and although limitations to this approach exist, using registered death information to supplement registry data reduces the probability of positively biasing survival estimates, and allows parametric modelling to proceed.
One advantage of predicting birth cohort median survival is that median survival can be estimated by gender and year of birth. Therefore, counselling parents of newly diagnosed patients on the prospect of their child's survival can be appropriately tailored. This is an improvement on current lifetable estimates, which are typically reported for an entire registry population. For example, the US CFF reported a current lifetable median survival estimate of 37.4 years in 2008,3 whereas model-derived birth cohort median predicted survival estimates suggest that US male and female patients born between 1985 and 1994 and surviving their first birthday live a median of 50.9 and 42.4 years respectively. As in others studies of CF survival which considered gender and birth cohort as explanatory factors, this study found that the period effect was the strongest determinant of survival into adulthood.19 Second, clinical management of patients with CF is complex and the survival benefit from novel therapies, early diagnosis and specialist, multi-disciplinary care remain unclear. Projecting birth cohort survivorship beyond the treatment period using a parametric approach could provide better insights into the effectiveness of current clinical practices.
A comparison of model estimates of median predicted survival between the 1980–1984 and 1985–1994 birth cohorts reaffirms that CF survival is improving in both the USA and Ireland. As in other studies, a gender gap in CF survival can be observed,20 21 however data comparing observed survival by gender across sequential birth cohorts are not often published,5 22 23 making it difficult to interpret our findings.
Differences in median survival estimates appear to exist internationally in this study and other studies.23 In addition to factors such as variability in diagnostic practices, genotype distribution and patient and transplantation management strategies between countries,24–26 the methodological approach used to estimate survival should be considered as a possible source of discordance during international survival estimate comparison. Cross-sectional comparison (ie, at a given time point) of international median predicted survival estimates derived using current lifetables has been shown to be misleading.27 Comparison of median age at death estimates derived from decedent CF populations has also been undertaken,24 but this measure of survival is considered unsatisfactory because it underestimates the observed median survival derived from following an entire CF population.27 Although this study has shown that parametric models can be fitted to registry datasets to derive estimates of median predicted survival, registry information has inherent limitations that can affect the reliability of model-derived survival estimates.
CF registries acknowledge that their datasets are not entirely representative of the CF population, and report population coverage levels of approximately 90%.20 28 Because patient consent is required for registry enrolment, registries are often unable to observe those who died before being invited to enrol, those opting not to participate, and others yet to be identified by the registry. To avoid the introduction of bias into birth cohort lifetables, all affected people need to be included. Differences in median survival estimates may be partly explained by potential biases in data collection by CF registries. Unlike the US datasets, national mortality records were included in the Irish birth cohort datasets, although deceased patients with CF in Ireland may not have been identified if CF was not reported as the underlying cause of death (eg, trauma, suicide), but rather a factor contributing to death. The omission of unrecognised deceased patients with CF from the US CFF Patient Registry prior to 1986, particularly childhood mortality data, which are not insubstantial,16 may have resulted in US survival estimates being positively biased. Although the large size of the US datasets allows more precise estimates of birth cohort lifetable predicted median survival to be derived, the effect of bias on survival estimation is not reduced.
Although the inclusion of additional explanatory variables (potentially available for prospectively followed birth cohorts) could further refine the predictive ability of this model, this was not possible for the analysis of the Irish data. Instead, our aim was to examine whether survival estimates could be projected beyond observed follow-up data in the first instance and identify a suitable model structure which could potentially be adopted by others.
Estimates of survival are important for planning service requirements and evaluating interventions, but in reality, how long any newborn baby with CF will live cannot be known. As with any model projecting survival estimates beyond observed data, population ascertainment, size and duration of follow-up of observed data affect the sensitivity of our model. When observed and predicted birth cohort lifetable median survival estimates were examined for our validation cohort, we found that estimates were comparable but also that the model was valid in a population smaller than that used to develop the model.
In conclusion, parametric models of birth cohorts can overcome the challenge of estimating birth cohort lifetable predicted median survival following a short follow-up period for a disorder experiencing continued improvement in survivorship. This approach may provide better opportunities to monitor survival and plan CF services at the local level.
Acknowledgments
We thank the UK and Australian CF registries for their willingness to contribute data to this analysis, but who were restricted in doing so by the absence of registry recorded deaths from their registries prior to 1998. We also are grateful to Professor Philip Farrell and Professor Chris Goss for critically reviewing an earlier draft.
References
Footnotes
In memoriam of Mrs Linda Foley, Chief Executive of the Cystic Fibrosis Registry of Ireland (2001–2009).
Funding This work was supported by the Cystic Fibrosis Registry of Ireland Executive Council. Health Research Board, Ireland (RP/2007/249).
Competing interests None declared.
Ethics approval Cystic fibrosis registry data were used in this study, and the registry was given ethical approval by each Ireland cystic fibrosis centre and clinic to collect data on cystic fibrosis patients.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves