Article Text

Abstract
Background In questionable cystic fibrosis (CF), mild or monosymptomatic phenotypes frequently cause diagnostic difficulties despite detailed algorithms. CF transmembrane conductance regulator (CFTR)-mediated ion transport can be studied ex vivo in rectal biopsies by intestinal current measurement (ICM).
Objectives To describe reference values and validate ICM for the diagnostic classification of questionable CF at all patient ages.
Methods ICM was performed in 309 rectal biopsies from 130 infants, children and adults including patients with known pancreatic-insufficient (PI)-CF (n=34), pancreatic-sufficient (PS)-CF (n=7), patients with an unclear diagnosis with mild CF symptoms, intermediate sweat test and/or CFTR mutation screening (n=61) and healthy controls (n=28). ICM was correlated to sweat chloride, extensive CFTR genotype and transcript analysis in the diagnostic group. The results were compared with previous ICM data in subjects with CF, congenital bilateral absence of the vas deferens, heterozygotes and controls.
Results The cumulative chloride secretory response of ΔIsc,carbachol, ΔIsc,cAMP/forskolin and ΔIsc,histamine was the best diagnostic ICM parameter (cut-off 34 μA/cm2 between patients with known PS-CF and controls), differentiating patients with questionable CF into PS-CF (n=6) and ‘CF unlikely’ (n=55) groups. Extensive genotype analysis detected two mutations (40% disease-causing) in 100% of individuals classified as PS-CF compared with 1.8% in those classified as ‘CF unlikely’.
Conclusions This comprehensive investigation of CFTR function and genotype underlines the diagnostic value of ICM, especially for confirmation of CF in the absence of two disease-causing CFTR mutations, exclusion of CF despite intermediate sweat test and age groups unsuitable for nasal potential difference measurements. ICM is an important tool for functional assessment in CFTR mutations of unknown clinical relevance.
- CFTR function
- sweat chloride
- diagnosis
- genotype
- algorithm
- cystic fibrosis
- paediatric lung disaese
Statistics from Altmetric.com
Introduction
Cystic fibrosis (CF) is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene.1 The clinical relevance in most of the >1500 described mutations is unknown.2 At present, only 23–28 of them are clearly accepted to be CF disease-causing based on functional CFTR characterisation2 3; others have been identified as neutral sequence variants.1 The wide range of CFTR mutation classes with different intracellular consequences on the CFTR protein basic defect and modifying genes lead to an enormous variability in the clinical CF phenotype.4
Various definitions of ‘milder’ CF forms have been proposed, including attempts to differentiate the wide spectrum of clinical CF phenotypes into ‘severe/mild’, ‘typical/atypical’ or ‘classic/non-classic’.4–8 In contrast, single-organ disease phenotypes with associated demonstration of CFTR gene abnormality but not fitting the current CF diagnostic criteria have been described as CFTR-related diseases.6 Diagnostic criteria for CF had been established9 but were often insufficient to exclude or confirm questionable ‘non-classic’ CF10–12 due to intermediate or normal sweat chloride (Cl−) results and lack of disease confirmation or exclusion by genetics. Nasal potential difference (NPD) measurements have been used to overcome diagnostic dilemmas,13 14 but they produce overlapping results in milder forms of CF and are unsuitable for use in infants and young children. In this complex situation, updated terminology and diagnostic algorithms3 5 15 16 have been suggested by consensus panels, resulting in a differentiation between the categories of CF (including pancreatic-insufficient (PI)-CF and pancreatic-sufficient (PS)-CF), ‘CF unlikely’ and an intermediate category (inconclusive/CF possible). However, the lack of adequate diagnostic classification and subsequent clinical care in a highly selected cohort of borderline cases remains a significant problem.
Ex vivo intestinal current measurements (ICM) have been used functionally to study the CFTR basic defect in human CF tissue,17–19 and have been shown to have potential diagnostic value. Mini-Ussing chambers are used to record the transepithelial short-circuit current (Isc) in freshly obtained human rectal suction biopsies as a measure of ion transport after stimulation with Cl− secretory agents. In this way, the CFTR Cl− channel, its amount of residual function and alternative Cl− channels can be investigated.20 21 This minimally-invasive safe procedure is applicable for all ages including newborn infants, it requires no sedation or special preparation and limitations are rare.21 To date, ICM has mainly been compared with NPD in cohorts of patients with CF and controls in research settings,22 23 so it is not included in the diagnostic algorithm and consensus criteria.3 5 16 However, experiences in patients with CF and controls have suggested promising sensitivity and specificity in the diagnosis of CF.
We therefore undertook a study to evaluate the diagnostic reliability of ICM in a large cohort of patients known to have CF, healthy controls and individuals with questionable CF presenting with mild symptoms and equivocal results in the standard diagnostic tests. We prospectively correlated a complete characterisation of the CFTR genotype with markers of CFTR Cl− channel function in a diagnostic cohort for the first time, and aimed to describe reference values and to validate the most informative ICM parameters.
Methods
Subjects
One hundred and thirty infants, children and adults (mean age 16.9±12.5 years; range 0.4–60.5; 23% <6 years; 35% ≥18 years; 54% male) were recruited at the Hannover CF centre and enrolled in the study between 1998 and 2009. This was a prospective study designed to evaluate the diagnostic accuracy of ICM (see also figure S1 in the online supplement), and diagnostic measurements remained unchanged over the whole study period. We investigated groups of known PI-CF (n=34), known PS-CF (n=7) and healthy adult volunteers (n=28) as disease and normal controls to determine the best diagnostic ICM cut-off value. As we are the reference centre for difficult CF diagnosis in Germany, patients with questionable CF (n=61) from all over Germany were sent to us and included subsequently in the study. Diagnosis in all known CF patients (PI+PS) had been established by typical CF symptoms plus either sweat Cl− concentration >60 mmol/l and/or CFTR mutation analysis according to agreed diagnostic criteria. All patients with an unclear diagnosis had a mild or monosymptomatic phenotype with sinopulmonary (nasal polyps, sinusitis, chronic cough/bronchitis, pneumonia, bronchiectasis, Pseudomonas aeruginosa airway colonisation), gastrointestinal (diarrhoea, failure to thrive, recurrent pancreatitis) or urogenital (azoospermia) symptoms compatible with ‘non-classic’ CF.6 8 Inflammatory rectal conditions such as those in ulcerative colitis were not present or clinically suspected in our cohort. Of the subjects with questionable CF, 92% presented with exocrine PS, verified by pancreatic stool elastase >200 μg/g. Sweat Cl− concentration after pilocarpine iontophoresis (38.2±23 mmol/l; two cases with insufficient sweat production) and/or CFTR mutation screening were equivocal, revealing one CFTR mutation in 26% of individuals. CFTR mutation screening was performed for the most common mutations according to local established laboratory techniques and patients' ethnic background (in 95% of individuals screening included ≥21 mutations). The reasons for further CF testing in cases with sweat Cl− <30 mmol/l and no CFTR mutation after screening were mostly uncertainties due to previously described CF cases with rare mutations in this sweat category and a strong desire by the families and caregivers to exclude CF.
Intestinal current measurement
Ion transport properties in all patients and healthy volunteers were studied by ICM in 309 rectal suction biopsies. The transepithelial Isc across the tissue was registered in recirculating Ussing chambers as described in detail previously.17 18 21 24 Briefly, superficial rectal suction biopsies were taken without sedation in a standardised procedure, mounted in Ussing chambers and incubated at 37°C with buffer solution. Basal potential difference (PDbasal), short-circuit current (Isc,basal) and transepithelial resistance (Rt,basal) were determined, and the Isc as a direct measure for the net movement of ions across the epithelium was recorded (usually for 60–75 min) after adding specific compounds to the mucosal (M) and/or serosal (S) bathing solutions: amiloride, indomethacin, carbachol, 8-bromo-cAMP and forskolin, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), histamine. A detailed description is given in the online supplement. Measurements of 2–4 biopsies were performed in all subjects. The mean individual results for basal tissue parameters and the maximal individual ΔIsc responses after stimulation with specific substances from all biopsies without technical problems (>98%) were used for further analysis. All ICM results were compared with sweat Cl− concentration.
Extensive CFTR genotype and transcript analysis
For a comprehensive characterisation of subjects and to confirm the diagnostic reliability of ICM, an extensive genetic analysis was performed in all 61 patients in the questionable CF group. Mutation screening of the entire coding sequences of the CFTR gene was performed from extracted genomic DNA using either CFTR sequencing or single-strand conformation polymorphism/heteroduplex analysis.25 DNA samples presenting with aberrant band patterns on either single or double strands were sequenced twice, in both directions. Moreover, the polymorphic sequence TGmTn in intron 826 (n=44) and a multiplex ligation dependent probe amplification (n=7) were determined in subgroups. Using RNA derived from nasal epithelial cells obtained by brushing, quantitative real-time RT-PCR was performed on a LightCycler 1.2 device to determine the amount of exon 9 skipping27 in the transcripts (n=19).
Statistical analysis
Data are presented as mean±SD unless otherwise indicated. Statistics were performed with SPSS 17.0 as appropriate. For comparison of ICM between PS-CF diagnostic and ‘CF unlikely’ groups, the Mann–Whitney U test was used because no normal distribution could be assumed and also because the sample size in the PS-CF group was small. All p values were two-tailed and p<0.05 was accepted as indicating statistical significance. The correlation between sweat test result and ICM was analysed separately for the ‘CF unlikely’ and CF (PS-CF diagnostic, PS-CF known and PI-CF known together) groups and for all CF subgroups individually. Because a linear relationship between the sweat test result and ICM could be assumed, the Pearson correlation coefficient was used.
Results
Intestinal current measurement: reference values
Reference data were obtained for the known PI-CF, PS-CF and healthy control groups. These results were compared with values from previous studies (PI-CF, n=240; PS-CF, n=20; congenital bilateral absence of the vas deferens (CBAVD), n=21; pancreatitis, n=7; heterozygotes, n=22; healthy controls, n=191) recorded with the same ICM set-up and protocol by the CF centres in Rotterdam and Hannover and were found to be very similar (table 1). In this way, a known clear cut-off value for ΔIsc,carbachol between subjects with PI-CF (<10 μA/cm2) and controls (>10 μA/cm2) was confirmed. Mean responses in subjects with PS-CF were higher than in those with PI-CF according to the gradient of CFTR dysfunction, and single PS-CF individuals showed high amounts of residual Cl− secretion with ΔIsc,carbachol responses up to 15 μA/cm2, representing about the lowest 5% of the range in controls. Obligate heterozygotes and controls were not distinguishable by ICM.
Reference values for ICM
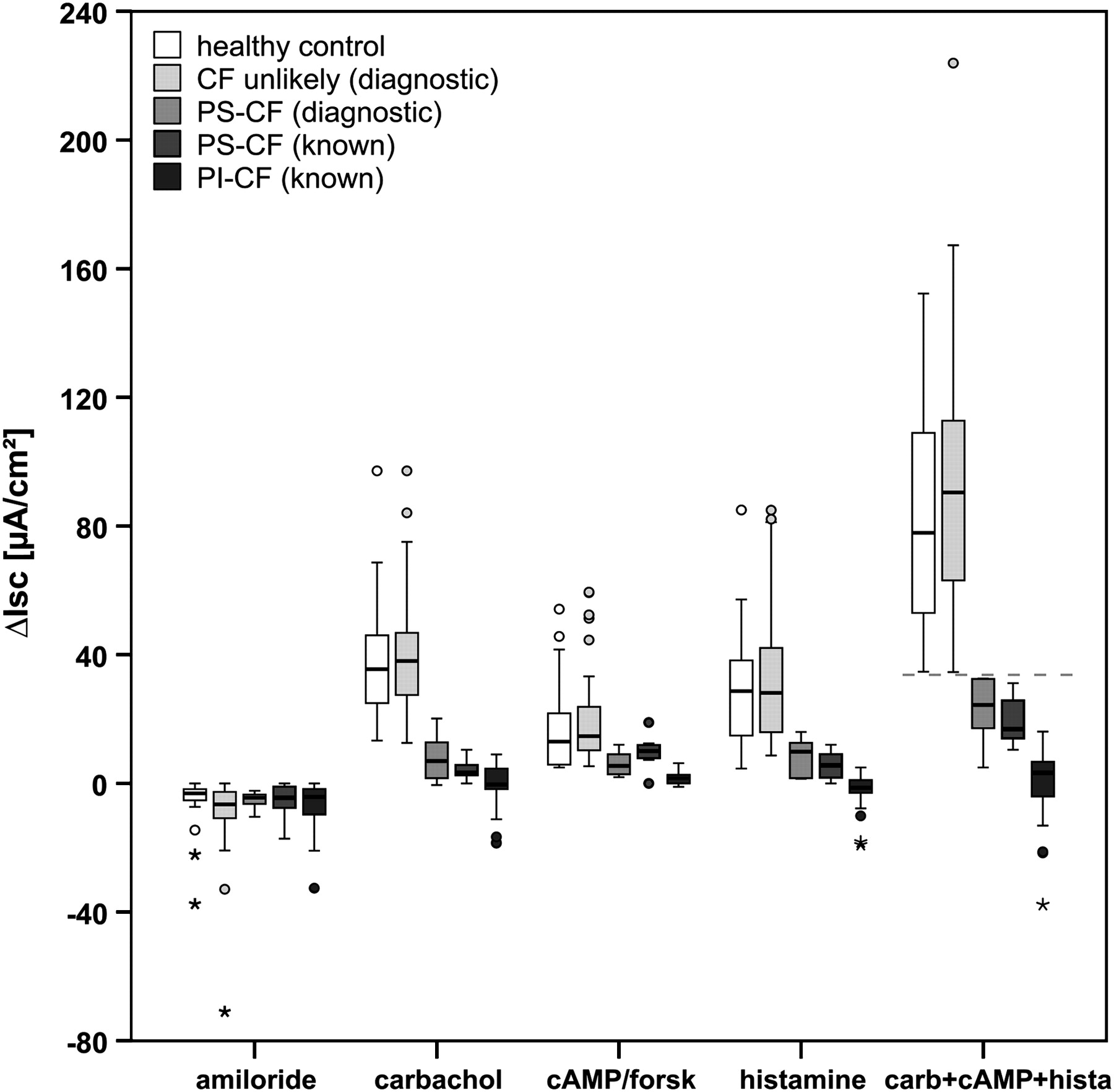
In our present cohorts of patients with known PI-CF, PS-CF and healthy controls, the best diagnostic ICM calculation was the cumulative value of the responses ΔIsc,carbachol, ΔIsc,cAMP/forskolin and ΔIsc,histamine (Isc,carb+cAMP+hista, taking into account both CFTR and alternative Cl− channels), with a clear cut-off value of 34 μA/cm2 between PS-CF and controls (figure 1); the maximal value in PI-CF was 16.1 μA/cm2. This new parameter provides the best diagnostic differentiation between patients with known PS-CF and controls, with 100% sensitivity and specificity in the present cohort with a previously known diagnosis (see figure S1 in online supplement).
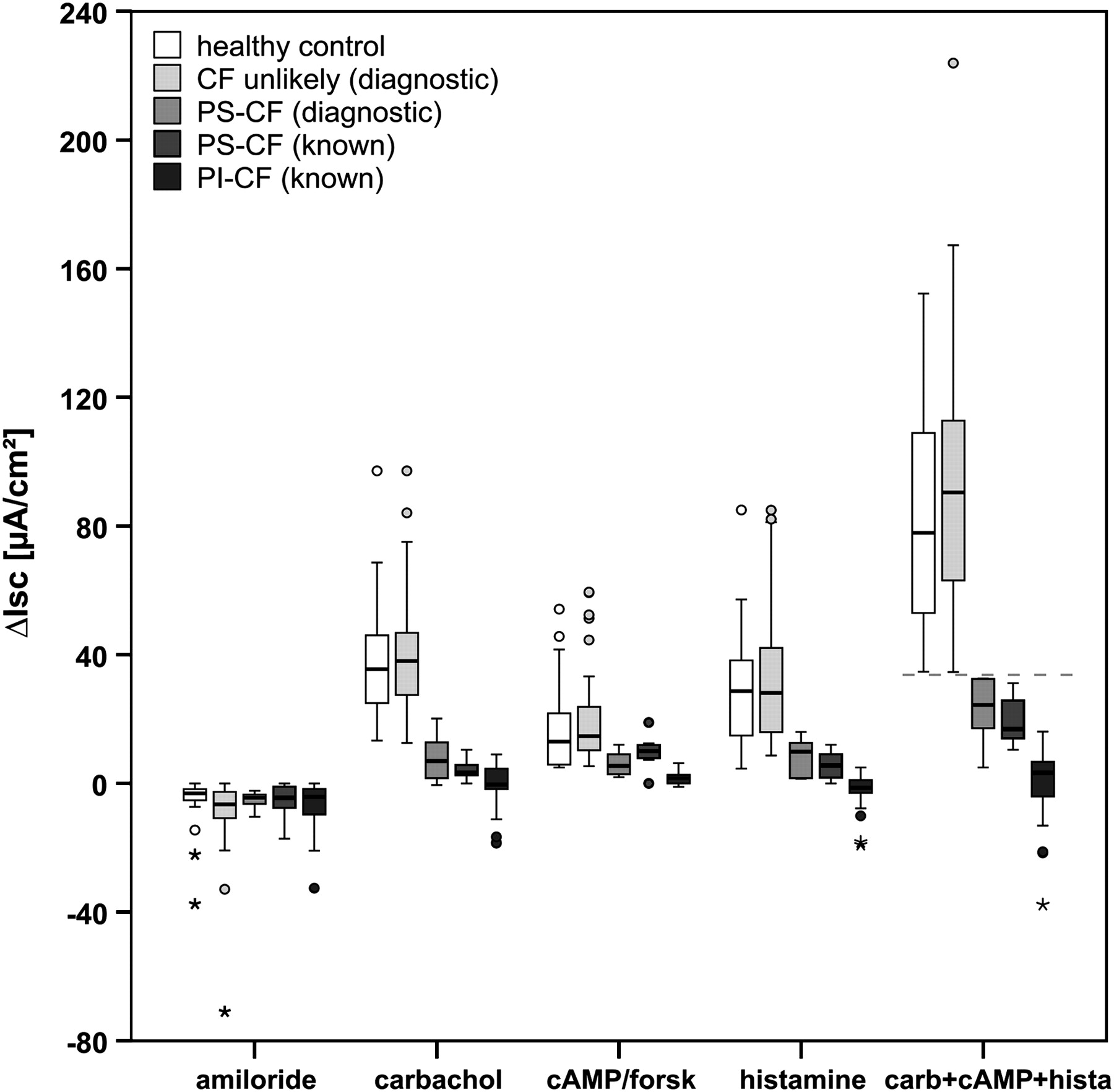
Reference values of ICM diagnostic parameter in groups of known PI-CF (n=34), known PS-CF (n=7), healthy controls (n=28) and validation in the present diagnostic cohort classified into PS-CF (n=6) and ‘CF unlikely’ (n=55) according to extensive CFTR genotype analysis and the previously determined ICM cut-off value of 34 μA/cm2 for Isc,carb+cAMP+hista (indicated by line). Changes in Isc after stimulation of rectal tissue with amiloride (100 μM), carbachol (100 μM), 8-Br-cAMP (1 mM)+forskolin (10 μM) and histamine (500 μM) are shown. Carb+cAMP+hista indicates the calculated individual cumulative ICM response of ΔIsc,carbachol, ΔIsc,cAMP/forskolin and ΔIsc,histamine. Boxplots indicate the IQR depicted by rectangle, with the upper horizontal line representing the upper quartile, followed by the median and the lower horizontal line representing the lower quartile. The range of non-outliers is given by the upper and lower whiskers initiating from the rectangle, whereby data points larger than upper quartile +1.5 IQR (or +3 IQR) and smaller than lower quartile −1.5 IQR (or −3 IQR) are considered outliers (indicated by circles) or extreme values (indicated by asterisks), respectively, shown as individual data points. CF, cystic fibrosis; ICM, intestinal current measurement; IQR, interquartile range; Isc, short-circuit current; PI, pancreatic-insufficient; PS, pancreatic-sufficient.
ICM in questionable CF: CFTR function, CFTR genotype and transcript analysis
On the basis of the newly established ICM reference data, the present diagnostic group with questionable CF was subsequently classified into PS-CF (n=6) and ‘CF unlikely’ (n=55) according to Isc,carb+cAMP+hista (the cumulative value of the responses ΔIsc,carbachol, ΔIsc,cAMP/forskolin and ΔIsc,histamine) with a cut-off at 34 μA/cm2 (figure 1).
The ICM basal tissue conditions in the diagnostic cohort corresponded to known values from subjects with PI-CF, PS-CF and healthy controls: Rt,basal 23.3±5.0 Ω×cm2 vs 23.3±5.5 Ω×cm2 (PS-CF vs CF unlikely, respectively; p=0.97), Isc,basal 44.7±37.8 μA/cm2 vs 57.4±35.9 μA/cm2 (p=0.22). After stimulation of the rectal tissue with different secretagogues, the net change in transepithelial Isc showed the following responses: ΔIsc,amiloride −5.2±2.9 vs –8.5±10.7 μA/cm2 (p=0.60); ΔIsc,carbachol 8.0±7.7 vs 40.9±18.1 μA/cm2 (p<0.001); ΔIsc,cAMP/forskolin 6.1±3.9 vs 19.5±13.4 μA/cm2 (p<0.001); ΔIsc,histamine 8.6±6.0 vs 32.4±19.7 μA/cm2 (p=0.001). For ΔIsc,carbachol, previously suggested as the best diagnostic parameter,21 in 2/6 subjects (33%) classified as PS-CF we obtained responses >10 μA/cm2, implying high residual Cl− secretory function, with one individual showing the highest response reported in PS-CF (20.2 μA/cm2). In contrast, 6/55 patients (11%) classified as ‘CF unlikely’ had a ΔIsc,carbachol response of <20 μA/cm2, demonstrating the limits of ΔIsc,carbachol as a diagnostic parameter. Correlation of ICM and sweat test results was performed in the ‘CF unlikely’ group, the CF group (PS-CF diagnostic, PS-CF known and PI-CF known together) and in all CF subgroups individually for ΔIsc,carbachol (figure 2A), ΔIsc,cAMP/forskolin (figure 2B), ΔIsc,histamine (figure 2C) and Isc,carb+cAMP+hista (figure 2D). The results are given in table 2. The correlation was consistently low for the ‘CF unlikely’ and PI-CF known groups and consistently moderate for the whole CF group. In the intermediate groups PS-CF diagnostic and PS-CF known, the calculation Isc,carb+cAMP+hista—newly established as the best diagnostic ICM marker—correlated with sweat Cl− concentration and confirmed the gradient of CFTR dysfunction while providing important diagnostic information in addition to the sweat test alone. The analysis revealed eight patients with questionable CF with sweat Cl− levels >60 mmol/l (normally compatible with a CF diagnosis if only based on sweat Cl−) and a normal ICM (Isc,carb+cAMP+hista, ΔIsc,carbachol), indicating organ-specific Cl− secretory function in a subgroup of individuals.

{kind=link}
{kind=link}
ICM and sweat chloride (Cl−) concentration results in patients with known PI-CF (n=25; only sweat sodium available in the others), known PS-CF (n=7) and diagnostic patients with questionable CF, classified into PS-CF (n=6) and ‘CF unlikely’ (n=53; no sweat test possible: n=2) according to cumulative ICM responses of ΔIsc,carbachol+ΔIsc,cAMP/forskolin+ΔIsc,histamine (Isc carb+cAMP+hista) and extensive CFTR genotype analysis. (A) ΔIsc,carbachol vs sweat Cl−; (B) ΔIsc,cAMP/forskolin vs sweat Cl−; (C) ΔIsc,histamine vs sweat Cl−; (D) Isc,carb+cAMP+hista vs sweat Cl−. Lines indicate the sweat Cl− categories normal (<30 mmol/l), intermediate (30–60 mmol/l) and pathological (>60 mmol/l) and the best ICM cut-off values for ΔIsc,carbachol (PI-CF <10 μA/cm2, PS-CF <20 μA/cm2, control >10 μA/cm2) and Isc,carb+cAMP+hista (PS-CF <34 μA/cm2, control >34 μA/cm2). CF, cystic fibrosis; ICM, intestinal current measurement; Isc, short-circuit current; PI, pancreatic-insufficient; PS, pancreatic-sufficient.
Correlation of ICM and sweat test results
Extensive genotype analysis detected the presence of two CFTR mutations (40% of which have been recommended to be CF disease-causing)2 3 in 100% of individuals classified by ICM as PS-CF compared with 1.8% of those classified as ‘CF unlikely’ (see table A in online supplement), supporting the diagnostic value of ICM. The functional consequence has not been explored before in many of the exhibited mutations. Genotype analysis in all individuals with sweat Cl− >60 mmol/l and normal ICM failed to detect a second CFTR mutation and we therefore classified them as ‘CF unlikely’. The T5 allele, which significantly influences the splicing efficacy of exon 9 and determines the amount of functional CFTR transcripts produced,28 was detected in nine ‘CF unlikely’ cases, six of whom has no other mutation found in the CFTR gene. Three patients with a T5 allele had one CFTR mutation (G551D/-; F508del/-; I177F/-), but TGm status (TG11) if available was suggestive to be benign, providing further evidence for the classification ‘CF unlikely’. A T5 allele with TG12 was present in two subjects with classification ‘CF unlikely’ (see table A in online supplement, ID 30 and 47), in which the possibility of a CFTR-related disease could not be ruled out at the present time. In the quantitative transcript analysis, the percentage of exon 9 skipping amounted to 18.0±12.5% (range 2.9–41.6%), with higher mean levels in patients with the T5 allele (29.6%) than in the others (12.6%), supporting the results of DNA analysis. Exon 9 skipping in healthy controls was 4.3–6.9% (mean 5.6%) (T7 and T9; n=4) and 34.0% (T5; n=1).
Diagnostic interpretation in all individuals, including those with pathological sweat tests, was made carefully, always based on individual clinical and CFTR phenotype. In all cases we informed patients and collaborating centres that a present-day classification as CF or ‘CF unlikely’ remains a clinical decision based on currently available diagnostic methods to characterise CFTR phenotype and genotype, and recommended a clinical long-term follow-up for all patients with pathological results in at least one diagnostic method.
Discussion
The diagnostic process for confirmation or exclusion of milder forms of CF is a clinical challenge with important implications for both patients and clinicians. Owing to heterogeneity of CFTR genotype and phenotype, the suspicion of CF arises in an increasing cohort of children and adults with mild possible CF symptoms which can lead to confusion, psychosocial consequences and misdiagnosis of CF despite detailed diagnostic criteria and algorithms. Sensitive methods in at least some expert centres that help to eliminate diagnostic dilemmas are therefore needed.
In this study we elucidated the diagnostic value of intestinal Cl− secretory function by ICM in rectal biopsies. The previously described definitions of CF are mainly based on clinical phenotype and sweat test and did not include diagnostic ICM data. However, the additional analysis of CFTR dysfunction by ICM in the present study is more complete, and a combination of methods characterising CFTR function in different organs (with possible different sensitivity to CFTR dysfunction) seems to be an essential diagnostic investigation of patients with a mild or monosymptomatic phenotype. Previous studies using NPD in questionable ‘non-classic’ CF confirmed the importance of characterising the degree of the CFTR basic defect in different organs.13 14 However, the limitation of using NPD in young children and nasal polyps is a relevant issue. An important advantage of the ICM method is its feasibility in the critical age group <6 years of age, starting at the time of newborn screening. We therefore included a large group of patients of all ages with questionable CF and investigated the reliability of ICM in comparison with sweat Cl− concentration and extensive CFTR genotype analysis for the first time.
For diagnostic interpretation, a previously suggested cut-off value for ΔIsc,carbachol as a single ICM parameter did not seem to be sufficiently reliable when being applied to the extreme phenotypes. Corresponding to sweat Cl− results in the borderline or normal range in patients with ‘non-classic’ CF, we report the first PS-CF individuals with ΔIsc,carbachol responses of up to 20 μA/cm2, indicating high residual CFTR function. The theoretical existence of rare patients with CFTR mutations resulting in less than 80–85% loss of CFTR function at the level of the colonocytes who escape detection by the ICM technique has been discussed previously.21 However, the results of this study suggest the cumulative response of ΔIsc,carbachol, ΔIsc,cAMP/forskolin and ΔIsc,histamine (Isc,carb+cAMP+hista) as the best diagnostic ICM parameter. In the relatively small cohorts of patients with known PI-CF, PS-CF and healthy controls used for validation of this parameter, Isc,carb+cAMP+hista showed sensitivity and specificity of 100%, supporting the evidence that ICM is currently the best diagnostic test for CF with no overlapping results between patients with PS-CF and controls. Although this study was designed to determine the diagnostic accuracy of ICM, the lack of a better gold standard than an extensive CFTR genotype analysis for the cohort with questionable CF might be a limitation, and the newly established ICM cut-off value should be applied to further patients referred for diagnostic investigation of questionable CF.
We are aware that even an extensive genotype investigation cannot function as a 100% diagnostic reference and that a sweat Cl− concentration >60 mmol/l in combination with possible CF symptoms, as in single patients classified as ‘CF unlikely’ by ICM and genotype analysis, would be compatible with a CF diagnosis. In contrast, the continuous gradient of CFTR dysfunction in CF and CFTR-related diseases, which is still under debate for terminology and diagnostic definitions, might also include single individuals with a false positive sweat test due to organ-specific differences in CFTR dysfunction. However, to our knowledge, the limitations of ICM in the diagnostic evaluation of questionable CF can be considered as minor, and the relatively simple exclusion of the diagnosis in the majority of individuals establishes the role of the procedure in the diagnostic algorithm for the future. Further comparisons with other markers of CFTR dysfunction such as sweat Cl− and NPD will enrich the discussion, and ICM in homozygous index cases can contribute to characterisation of the functional consequences of rare mutations on the CFTR basic defect and the clinical disease.23 29
In summary, we have established reference values and demonstrated that ICM is an elegant and reliable method in the diagnostic investigation of patients of all ages with mild or monosymptomatic CF phenotype and equivocal standard tests, even in cases of rare CFTR mutations with associated residual Cl− secretion. ICM provides an important diagnostic advantage for these otherwise inconclusive subjects and is an important tool—especially in the era of screening of newborn infants for CF—for functional CFTR assessment in patients with CFTR mutations of unknown clinical relevance.
Acknowledgments
The authors thank all patients, families and collaborating CF centres for their participation and help in this study, and H de Jonge and I Bronsveld for good collaboration and helpful discussions.
References
Supplementary materials
Web Only Data thx.2009.125088
Files in this Data Supplement:
Footnotes
Funding Deutsche Forschungsgemeinschaft SFB 621, C7 and the Swiss National Foundation (3200-066767.01, 310000-112652).
Competing interests None.
Ethics approval This study was conducted with the approval of the local ethics committee, MH Hannover, Germany and all patients and/or parents and healthy controls gave their written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.