Article Text

Abstract
Background and aim: Aberrant angiogenesis and defective epithelial repair are key features of idiopathic pulmonary fibrosis (IPF). Endostatin is an antiangiogenic peptide with known effects on endothelial cells. This study aimed to establish the levels of endostatin in the bronchoalveolar lavage fluid (BALF) in IPF and to investigate its actions on distal lung epithelial cells (DLEC) and primary type II cells.
Methods: 20 patients with IPF and 10 controls underwent BAL. Endostatin was measured by ELISA. BALF cytokines and matrix metalloproteinase (MMP)-3 were measured by Luminex array. Primary DLEC monolayers were wounded and treated with endostatin. Apoptosis and cell viability were assessed.
Results: Endostatin was elevated in the BALF and plasma of patients with IPF compared with normal controls. There was a negative correlation between endostatin, forced vital capacity and gas transfer. Endostatin correlated with a number of proinflammatory cytokines and MMP3. Physiological endostatin doses inhibited DLEC wound repair by 44% in an effect that was partially FasL and caspase dependent. Endostatin increased apoptosis rates by 8% and reduced their viability by 34%. Similar effects of endostatin were seen in primary type II cells in terms of inhibition of wound repair and proliferation.
Conclusions: Elevated BALF endostatin levels correlated with a number of elevated cytokines, MMP3 and lung function in IPF. Endostatin is a novel inhibitor of DLEC wound repair, inducing apoptosis and reducing cell viability in a FasL and caspase dependent manner. Endostatin may play a role in aberrant epithelial repair in IPF.
Statistics from Altmetric.com
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease of unknown aetiology.1 Current theories speculate that IPF results from abnormal wound healing in response to multiple microscopic sites of alveolar epithelial cell (AEC) injury, activation and apoptosis.2 AEC apoptosis has been implicated in the initiation of fibrotic foci3 4 and an increased rate of apoptosis could contribute to the loss of balance in cell turnover and abnormal re-epithelialisation. Caspase inhibition blocks AEC apoptosis and reduces the accumulation of collagen matrix in animal models.5
Collagens are a family of extracellular matrix proteins that play a dominant role in maintaining the tissue structural integrity. Excessive collagen deposition leads to fibrosis disrupting the normal functioning of surrounding tissues.6 Collagen XVIII is a major proteoglycan found in alveolar capillary and epithelial basement membranes. Proteolytic enzymes cleave peptide bonds within the protease sensitive hinge region of collagen XVIII to release anti-angiogenic endostatin fragments varying in size between 20 and 30 kDa.7 Endostatin has been implicated in the microvascular damage found in pre-eclampsia and can be used to suppress tumour neovascularisation/angiogenesis in mice.8 Endostatin levels have previously been found to be elevated in the serum of patients with IPF.9
Endostatin inhibits proliferation and migration of endothelial cells by interruption of focal adhesions and actin stress fibres, essential to cell motility.10 Endostatin induces endothelial cell apoptosis11 and has been shown to associate with α5β1 integrin, essential for endothelial cell proliferation and apoptosis,12 and vascular endothelial growth factor receptor 2 (VEGFR-2).13 Given the importance of the epithelium in IPF and the fact that AEC express both α5β1 integrin14 and VEGFR-2,15 we hypothesised that endostatin may have a role in inhibiting AEC repair in patients with IPF.
Matrix metalloproteinases (MMP) are essential for extracellular matrix remodelling, wound repair and angiogenesis, and have been implicated in the pathogenesis of IPF. MMP3 (stromelysin-1) has a wide range of actions influencing fibrinolysis and angiogenesis,16 and is able to cleave endostatin from collagen XVIII.17 Establishing MMP3 levels would give an indication of the potential for local proteolytic cleavage of endostatin.
The relationship between endostatin and cytokine networks is undetermined, but it is known that endostatin is able to alter expression of a wide range of genes.18 CXC cytokines that contain the terminal glu-leu-arg (ELR+ve) motif are potent induces of angiogenesis. Cytokines that do not present this motif (ELR−ve) which are mainly interferon (IFN) inducible are, in contrast, antiangiogenic. An imbalance in pro and antiangiogenic CXC cytokines has been described in IPF.19
The study aims were to determine bronchoalveolar lavage fluid (BALF) and plasma endostatin levels in IPF and then relate endostatin to lung function and alveolar cytokine levels. In vitro experiments were designed to establish whether physiological levels of endostatin can influence human primary distal lung epithelial cell actions and propose a potential mechanism of action.
METHODS
Subjects
Twenty patients with IPF diagnosed according to current American Thoracic Society (ATS) criteria were recruited from the specialist interstitial lung disease clinic at University Hospital Birmingham, UK. Open lung biopsy confirmed usual interstitial pneumonia in five patients where diagnosis was uncertain. Bronchoscopy was performed during the investigation stage after referral, before the start of definitive treatment. Ten healthy individuals free from respiratory disease were recruited as controls. This study was approved by the local ethics committee and patients gave written informed consent.
Measurements
Patients underwent bronchoscopy and bronchoalveolar lavage (BAL), as described previously.20 Endostatin was measured in BALF and plasma by an ELISA kit (R&D, Abingdon, UK) according to the manufacturer’s instruction. BALF cytokines (interleukin (IL)-8, epithelial neutrophil activating peptide-78 (ENA-78), IL1RA, IL6, monocyte chemoattractant protein-1 (MCP-1), IFNγ) and MMP3 were measured by Luminex array (R&D Systems).
Protein was measured using the Bio-Rad DC protein assay kit II. The protein permeability index was calculated as the ratio of BALF to plasma protein, as described previously.21
Pulmonary function testing
Forced vital capacity (FVC) was measured using the Jaeger Compact System (Viasys Healthcare, Basingstoke, UK). Total lung diffusing capacity for carbon monoxide (TLco) was measured by the single breath technique (Jaeger Compact System). Results were expressed as per cent of predicted values.
Cell culture
Primary human distal lung epithelial cells (DLEC) (Cambrex, Wokingham, Berkshire, UK) were cultured in complete growth media (SAGM, Cambrex), according to the manufacturer’s protocol. Cells were obtained from three separate donors and experiments were performed before passage 3. Confluent DLEC monolayers were wounded with a 2 mm mechanical wound, as described previously.22 The wounds were photographed under a microscope at 0 and 18 h and analysed with Scion image software.
A dose–response of 0–1000 ng/ml of endostatin (Molecular Probes, Poortgebouw, Leiden, The Netherlands) on DLEC wound repair was assessed. Experiments were then repeated by incubating endostatin alone or preincubating with anti-FasL antibody (1 ng/ml) (R&D) or the caspase inhibitor Z-DEVD-FMK (20 μM) (R&D).
Actions of endostatin on DLEC apoptosis and cell viability
DLEC apoptosis was assessed using annexin/propidium iodide fluorochromes (Invitrogen, Loughborough, UK) and analysed by flow cytometer (Coulter EPIC flowcytometer) and Cellquest software. This experiment was repeated with anti-FasL antibody or after preincubating the monolayer with the caspase inhibitor Z-DEVD-FMK, as above. Cellular viability was measured using cell titre (Promega, Southampton, UK), according to the manufacturer’s instructions.
Alveolar type II cell extraction and culture
In order to assess the effects of endostatin on ATII cells, lung samples were obtained from four patients undergoing lung resection for lung cancer. ATII cells were extracted according to the method of Witherden and Tetley.23 Average yields of ATII were 30 million cells per resection with a purity of 92%. Cells were tested for ATII cell phenotype by alkaline phosphatase staining, lysotracker lamellar body staining and by PCR expression of surfactant protein C—a type II cell marker with negative expression of aquaporin V (a type I cell marker) (data not shown). For viability experiments, cells were used 24 h after extraction. For wound repair experiments, 0.5 million cells were seeded onto 24 well plates and grown for 4 days in DCCM (Troon Scientific, Troon, Perthshire, UK) media supplemented with 10% fetal calf serum. Wound repair assays were performed as per the DLEC methods above. Bromodeoxyuridine (BRDU) incorporation of ATII cells was assessed using a colorimetric assay (Calbiochem, UK) according to the manufacturer’s instructions. To allow for variability of basal proliferation between batches of ATII cells, cells were stimulated with 10 ng/ml of tumour necrosis factor α for 1 h prior to addition of BRDU and results expressed as per cent control BRDU incorporation.
Statistics
Normally distributed data, assessed by the Komolgorov–Smirnoff test, is presented as mean (SE). Comparison between the two groups was analysed using the Student’s t test and multiple groups by one way ANOVA with Tukey’s post hoc analysis. Non-parametric data are presented as median and interquartile range (IQR). Between group comparisons were performed using the Mann–Whitney U test; multiple groups using the Kruskal–Wallis test. The cytokine tests had a Bonferroni correction applied; a p value <0.005, as opposed to p⩽0.05 for all other data, was considered significant. Correlations were made using Spearman rank. This study was considered hypothesis generating so a power calculation was not performed. Statistics were performed using SPSS 15.
RESULTS
Demographics
Twenty patients with IPF and 10 healthy controls were recruited. A summary of the patient characteristics is shown in table 1. Six patients agreed to a sequential BAL 6 months after starting treatment with prednisolone and azathioprine with or without N-acetylcysteine. The mean age of the IPF patients was higher than healthy controls but there was no relationship between BALF endostatin and age (rho = −0.161, p = 0.443).
BALF and plasma levels of endostatin are elevated in patients with IPF
Endostatin was detectable in the BALF and plasma of all patients and normal control samples. Endostatin was significantly elevated in both BALF (IPF mean 1.32 (SE 0.20) ng/ml, normal 0.12 (SE 0.03) ng/ml; p<0.001) and plasma (IPF mean 259.2 (SE 41.3) ng/ml, normal 94.7 (SE 18.9) ng/ml; p<0.001) of IPF patients compared with controls (fig 1A). Plasma endostatin levels correlated with BALF endostatin (r = 0.336, p = 0.007). Endostatin levels remained elevated in the six patients who underwent sequential BAL after treatment; first BALF average endostatin 1.02 (SE 0.29) ng/ml, second BALF 1.42 (SE 0.29) ng/ml (p = 0.792) (data not shown).

BALF endostatin correlates with markers of lung function
BALF endostatin negatively correlated with FVC (r = −0.604, p = 0.006) and TLco (r = −0.612, p = 0.005). BALF neutrophil per cent also correlated with FVC (r = −0.627, p = 0.004) and Kco (r = −0.528, p = 0.02). BALF endostatin correlated with BALF protein (r = 0.740 p = 0.001) and the protein permeability index (r = 0.396, p = 0.05).
BALF cytokine levels
A number of BALF cytokines were elevated compared with normal controls, including the ELR+ve cytokines ENA-78 and IL-8 (table 2).
Cytokines were measured using a luminex array. Results were compared by Mann–Whitney U test between patients with IPF and healthy controls. After Bonferroni correction, a p value <0.005 was considered significant.
Correlations between BALF endostatin and cytokines
BALF endostatin correlated with the ELR+ve cytokines IL-8 (rho 0.562, p<0.001) and ENA-78 (rho = 0.501, p = 0.008), neutrophil per cent (rho = 0.508, p = 0.001) and the cytokines IL1ra (rho = 0.726, p<0.001), MCP-1 (rho = 0.512, p = 0.005) and IL-6 (rho = 0.607, p = 0.008).
BALF levels of MMP3 are elevated in IPF and correlate with BALF endostatin
Median IPF BALF MMP3 levels were 24.8 pg/ml (IQR 32.2) compared with 9.71 pg/ml (IQR 5.2) in normal individuals (p = 0.009) (fig 1B). BALF endostatin correlated with BALF MMP3, supporting a role for MMP3 in endostatin release from type XVIII collagen in IPF (rho = 0.45, p = 0.033) (data not shown). MMP3 levels also correlated with neutrophil percentage (r = 0.424, p = 0.039), IL8 (rho = 0.625, p = 0.001) and granulocyte-colony stimulating factor (rho = 0.599, p = 0.003). There was no relationship with ENA-78 or IFNγ.
Endostatin effects on DLEC wound repair, apoptosis and cell viability
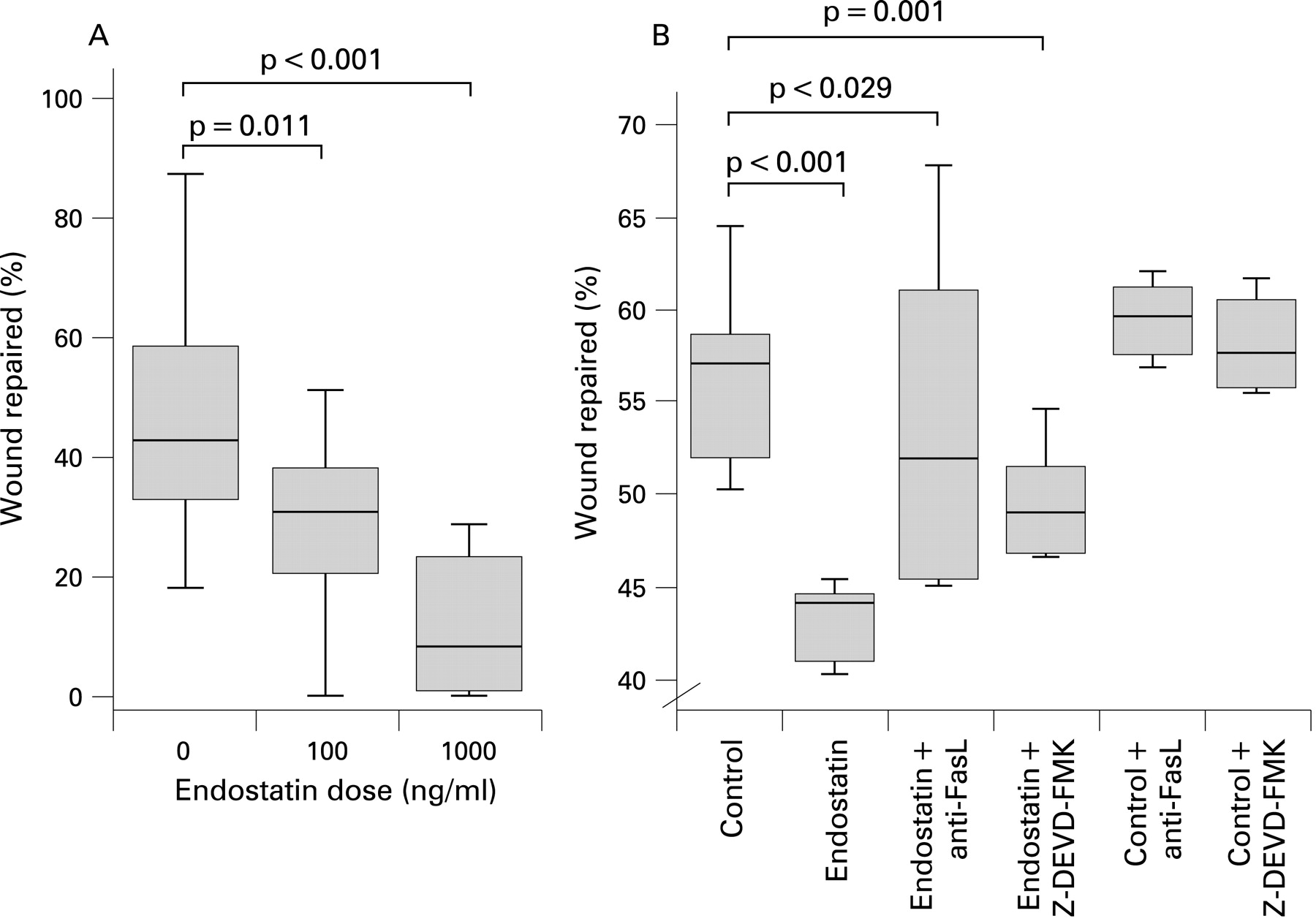
Endostatin inhibited wound repair in DLECs (fig 2A) and this effect was partially blocked by FasL antibody and the caspase inhibitor Z-DEVD-FMK; control (56.58% (SE 2.1)), endostatin 100 ng (42.52% (SE 0.65)), endostatin+anti-FasL antibody (54.21% (SE 4.44)) and endostatin+Z-DEVD-FMK (49.2% (SE 0.79)), ANOVA p<0.001). There was a significant difference between control and endostatin (p<0.0001), between endostatin and endostatin+anti-FasL antibody (p = 0.029) and endostatin+Z-DEVD-FMK (p = 0.001) (fig 2B). At higher endostatin doses, endostatin often increased the measured wound which is described as a negative percentage wound repaired.

Apoptosis rates were higher in endostatin treated cells than controls (100 ng 58.8% (SE 0.98), 1000 ng 58.9% (SE 0.81), controls 50.8% (SE 1.21), ANOVA p<0.001 and Tukey’s p<0.001, p<0.001, respectively) (fig 3A). The affect of endostatin on DLEC apoptosis was attenuated by anti-FasL antibody (mean 56.9% (1.0) vs 45.7% (4.4), p = 0.024). Caspase inhibition reduced the endostatin effect but this did not achieve statistical significance (mean 56.9% (1.0) vs 50.4% (1.3), p = 0.289) (data not shown).

Cell titre colorimetric assay showed reduced cell viability in endostatin treated DLEC cells and that this effect was enhanced at a higher dose (p<0.001) (fig 3B).
Effect of endostatin on primary human ATII cell wound repair and proliferation
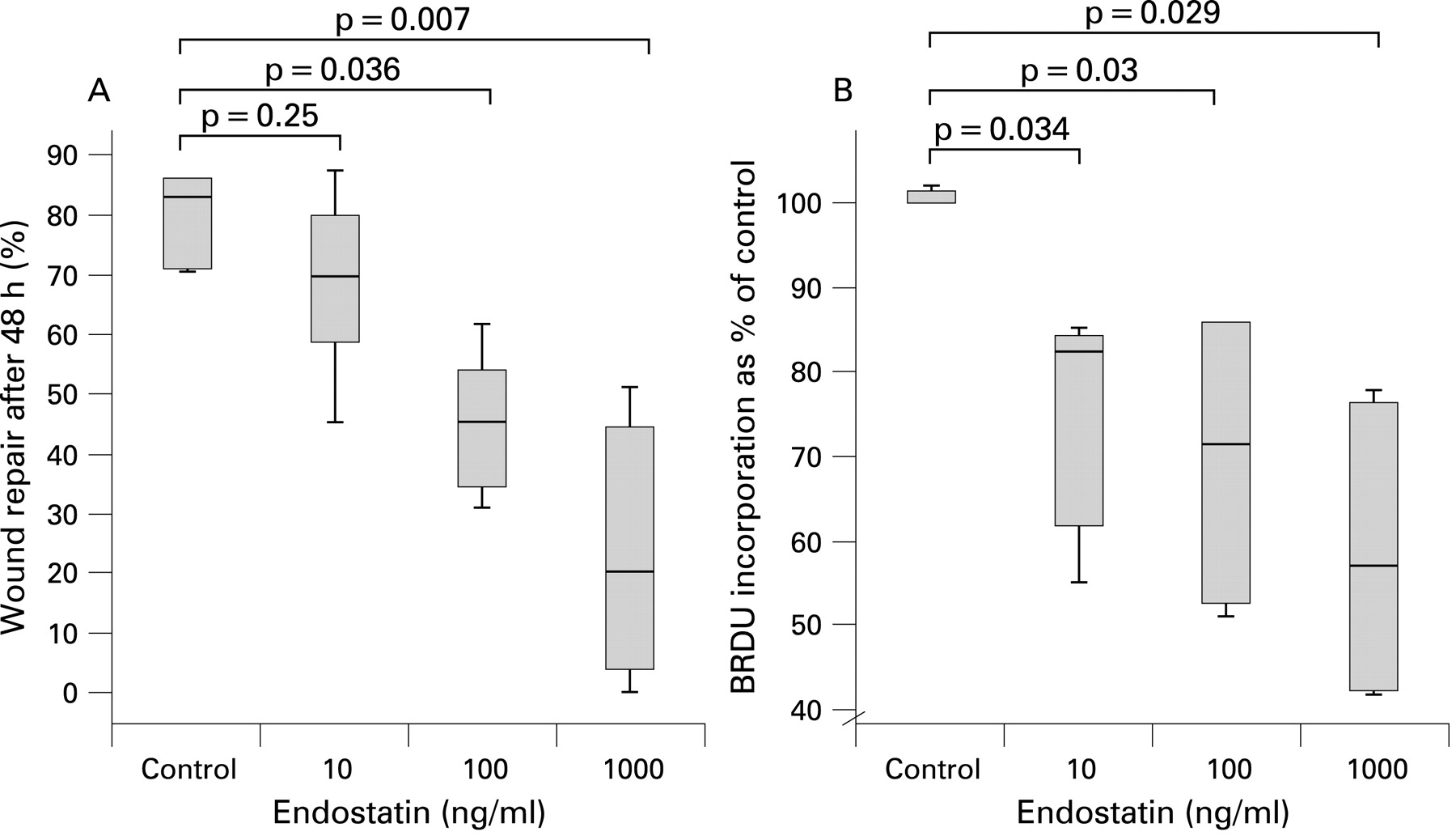
Endostatin caused a dose dependent reduction in wound repair after 48 h (fig 4A). Unlike in DLEC, endostatin did not reduce cellular viability over 24 h, suggesting that in ATII this action was not associated with cell death (data not shown). To confirm that the effects of endostatin on wound repair were related to proliferation, we assessed ATII cell BRDU incorporation. Endostatin inhibited BRDU incorporation in a dose dependent manner over 36 h (control (100% IQR 100–101.42), endostatin 10 ng/ml (82.5% (IQR 61.75–84.5, p = 0.034), endostatin 100 ng/ml (71.5% (IQR 52.5–86), p = 0.03) and endostatin 1000 ng/ml (57% control incorporation (IQR 42.25–76.25), p = 0.029) (fig 4B)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
This is the first study to demonstrate endostatin in the alveolar space of patients with IPF and that levels are elevated compared with normal controls. The relationship between BALF endostatin and physiological severity indicates a potentially important clinical role for endostatin in IPF. In addition to its known inhibitory effects on endothelial cells, this study confirms actions of endostatin on DLEC and ATII wound repair. These effects on DLEC appear to involve apoptosis, be partially FasL dependent and can be abrogated by caspase inhibition.
Two forms of collagen XVIII are expressed in normal human tissues, designated the SHORT and LONG variants.24 The LONG form appears to be largely produced by hepatocytes in the liver sinusoids and is measurable as a plasma protein. The SHORT form is expressed in most vascular and epithelial basement membranes, including the specialised capillaries found in lung alveoli. The carboxy terminus of type XVIII collagen includes a hinge region that displays protease sensitive sites which generate the endostatin fragments. A number of enzymes, including neutrophil elastase, cathepsins and matrix metalloproteinases (including the fibroblast derived MMP3) have been implicated in the pathogenesis of IPF.6 These proteases are known to act on this hinge region to release not only endostatin but other larger fragments that contain the endostatin fragment. Previous studies have suggested that these C terminal fragments may have similar antiangiogenic actions to endostatin.17
Endostatin levels in the IPF lung may be elevated because of increased local cleavage of alveolar collagen XVIII. Gene expression array data suggest an increase in type XVIII collagen mRNA in whole lung extracts from patients with IPF,25 and in an animal model of FasL induced lung injury there is upregulation of type XVIII collagen mRNA.26 Although our correlations between MMP-3 and endostatin do not prove a mechanistic link, taken together with our findings of increased BALF MMP-3 in IPF, these data suggest that local intra-alveolar degradation of type XVIII collagen may be important in endostatin generation. The proteolytic cleavage of collagen XVIII may only partially govern endostatin levels as it has been shown that endothelial endostatin release may be induced by general cell stress and can be modulated by the nitric oxide/cGMP pathway.27 A wide range of inflammatory cytokines were measured in this study and found to correlate with endostatin suggesting endostatin generation may be upregulated by an alveolar inflammatory milieu. Alternatively, the observed relationship between BALF endostatin and protein permeability, and the plasma:BALF endostatin gradient, suggests that alveolar levels of endostatin may be raised as a result of leakage from the microvasculature into the alveolus at sites of increased alveolar barrier permeability. Given the antiendothelial cell actions of endostatin, endostatin may play a pathophysiological role in this abnormal permeability.
Prior to this study the main focus of research into the biological effects of endostatin was on endothelial cells in angiogenesis and cancer biology. The actions of endostatin on endothelial cells has variously been shown to reduce migration, spreading and induce apoptosis.28 29 Not all studies have confirmed these effects and often these experiments have used supraphysiological doses. This study described the actions of endostatin on epithelial cells for the first time. Physiological doses of endostatin significantly reduced both DLEC and ATII cell wound repair with inhibitory effects on cellular viability and increased apoptosis in DLEC. These actions on DLEC were partially mediated by FasL and caspase pathways; this is relevant as FasL induced epithelial apoptosis has been implicated in the pathogenesis of IPF. Endostatin is known to promote the formation of lipid rafts essential for signalling through the Fas death receptor30 and provides a potential mechanism whereby endostatin induces epithelial apoptosis in our patients. The relatively small increase in DLEC apoptosis (fig 3A) caused by endostatin suggests additional mechanisms may be involved in endostatin inhibition of DLEC wound repair. Spreading and migratory processes are essential for monolayer repair, and the effects of endostatin on these mechanisms warrant further investigation.
It has been suggested that disordered epithelial remodelling promotes fibroproliferation in IPF. Endostatin can interact with both integrins and VEGFR-2 which are known to be present on epithelial cells.12 The known effects of endostatin on WNT and cyclin D1 expression may be important in its epithelial effects as pulmonary epithelial cell turnover is also regulated by these signalling pathways.31 Further work to characterise the mechanism of endostatin action on epithelial cells is required. Nevertheless, our study suggests that there may be a direct link between collagen degradation and ongoing epithelial cell apoptosis induced by endostatin production in patients with IPF.
This study has several limitations. Firstly, our patient population did not all have lung biopsies to prove usual interstitial pneumonia although they were well characterised according to international guidelines in a specialist clinic. Furthermore, the difference in age between the patients with IPF and our healthy controls may be a confounding factor when comparing endostatin levels. To address this we examined the relationship between age and endostatin levels in IPF and normal controls and found no correlation. Finally, we are unable to assess the relative importance of endostatin within BALF in an angiogenesis bioassay because of the current lack of an effective inhibitor of endostatin bioactivity.
In conclusion, this study has demonstrated elevated endostatin levels in IPF patients. Endostatin levels correlated with the degree of lung function impairment and levels of inflammatory mediators that are associated with angiogenesis. Endostatin has been found to inhibit primary epithelial cell wound repair by mechanisms that involve both increased apoptosis rates or inhibition of proliferation. In conjunction with its antiangiogenic properties therefore, the presence of endostatin within the lung in IPF may result in defective alveolar epithelial repair. This study raises the possibility of an endostatin inhibitor as a therapeutic option in IPF.
Acknowledgments
We would like to thank the staff of the Birmingham Wellcome Clinical Research Facility.
REFERENCES
Footnotes
Funding: AR was funded by a Wellcome CRF Entry Level Fellowship. DRT is funded by a Wellcome Intermediate Fellowship.
Competing interests: None.
Ethics approval: This study was approved by the local ethics committee.