Article Text

Abstract
The main risk factors for ventilator induced lung injury are reviewed, together with the possible mechanisms and its clinical relevance.
- ventilator induced lung injury
Statistics from Altmetric.com
Mechanical ventilation has become an indispensable tool, facilitating general anaesthesia and supporting life in the critically ill. However, its application has adverse effects including an increased risk of pneumonia, impaired cardiac performance, and neuromuscular problems relating to sedation and muscle relaxants. Moreover, it has become clear that applying pressure—whether positive or negative—to the lung can cause damage known as ventilator induced lung injury (VILI). This concept is not new. In his treatise on resuscitation of the apparently dead, John Fothergill in 1745 suggested that mouth to mouth inflation of the victim's lungs might be preferable to using a pair of bellows as “the lungs of one man may bear, without injury, as great a force as another man can exert; which by the bellows cannot always be determin'd”.1 Although not specifically addressed, Forthergill's admonition against the use of the bellows probably related to gross air leaks produced by large pressures. This type of injury is now called barotrauma and was the first widely recognised manifestation of VILI. The clinical and radiological manifestations of barotrauma include pneumothorax, pneumomediastinum, and surgical emphysema.
Later, evidence accumulated to suggest that ventilation causes more subtle morphological and functional changes and can excite an inflammatory response within the lung. This type of injury was not recognised for many years as the pattern of damage is often indistinguishable from that seen in other forms of lung injury such as the acute respiratory distress syndrome (ARDS), for which mechanical ventilation is an indispensable treatment. Studies using animal models were necessary to define key aspects of VILI. Based on these, many clinicians in the 1990s began to adopt ventilatory strategies designed to minimise lung injury, although the clinical importance of VILI has only recently been established.2
In this review we summarise the main risk factors for VILI, its possible mechanisms and its clinical relevance. Specific ventilation techniques for ARDS are addressed in a separate article in this series3 and will only be alluded to here for the purpose of illustrating general principles.
MAJOR DETERMINANTS OF VILI
Most research into VILI is based on positive pressure ventilation delivered via an endotracheal tube, although the principles are equally relevant to non-invasive or negative pressure ventilation. It has become clear that the degree of VILI is determined by the interaction of the ventilator settings and patient related factors, particularly the condition of the ventilated lung.
Ventilator determinants of VILI
Airway pressure and lung distension
Conceptually, it seems obvious that inflation of the lung will cause damage if airway pressures are high enough. The important issues for the clinician have been (1) what levels of airway pressure are dangerous and (2) can they be avoided in the mechanical ventilation of patients with stiff lungs, as in ARDS?
The association between high airway pressure and air leaks has long been recognised.4,5 However, this relationship does not necessarily imply causality as damaged, stiff lungs that require high airway pressures for ventilation may be intrinsically more prone to air leaks. Recent large studies in patients with ARDS have, in fact, shown a poor correlation between airway pressure (or tidal volume) and the occurrence of air leaks, which occurred in 8–14% of the patients.2,6–8 However, these data should not be interpreted as demonstrating that the degree of lung distension is unimportant in the development of barotrauma. In these studies airway pressures and tidal volumes were lower than those used in the past when barotrauma rates as high as 39% were reported in patients ventilated for acute respiratory failure.9
More subtle lung damage was first unequivocally shown by Webb and Tierney who ventilated rats for 1 hour using different airway pressures with and without positive end expiratory pressure (PEEP).10 Animals ventilated with a peak airway pressure of 14 cm H2O had no histological changes in the lung, while those ventilated with a pressure of 30 cm H2O had mild perivascular oedema. In contrast, all rats ventilated at 45 cm H2O (without PEEP) developed severe hypoxia and died before the end of the hour. Their lungs had marked perivascular and alveolar oedema. Similar findings have been observed in other species, although there is considerable variation in the susceptibility to VILI. Whereas in rats ventilation at high peak inspiratory pressure for just 2 minutes is sufficient to induce pulmonary oedema, larger animals such as rabbits and sheep require much longer periods of ventilation for changes to be evident.11–13 This is clearly important when extrapolating data from animal studies to humans.
Pressure or volume?
Although the term barotrauma is commonly used when discussing VILI, the evidence indicates that the degree of lung inflation is a more important determinant of lung injury than airway pressure per se. This may be inferred from the observation that trumpet players commonly achieve airway pressures of 150 cm H2O without developing repetitive episodes of gross air leakage.14 The relative contribution of pressure and volume to lung injury was first studied by ventilating rats whose tidal excursion was limited by strapping the chest and abdomen.15 High airway pressure without a high tidal volume did not produce lung injury. By contrast, animals ventilated without thoracic restriction using high tidal volumes, achieved either with high positive inspiratory pressure or negative pressure in an iron lung, developed severe injury. These results have been confirmed in other species.16,17
The term volutrauma is therefore more accurate than barotrauma, although in practice the two are closely related. The degree of alveolar distension is determined by the pressure gradient across the alveoli, approximated by the transpulmonary pressure, the difference between the static airway pressure (estimated clinically by the plateau airway pressure) and the pleural pressure. Peak airway pressure is not necessarily a reflection of alveolar pressure and is greatly influenced by the resistance to flow in the airways. Recent guidelines have therefore emphasised the importance of limiting plateau pressures and of being aware of factors that increase (or decrease) the degree of alveolar distension for a given alveolar pressure.18 For example, conditions associated with increased chest wall compliance such as immaturity increase lung distension, and hence damage, for a given alveolar pressure. Conversely, chest wall compliance is commonly reduced by abdominal distension,19 which can result in lower alveolar inflation, derecruitment, and hypoxaemia if the plateau pressure is inappropriately low.
Lung injury at low lung volumes
There is evidence that lung damage may also be caused by ventilation at low lung volume (meaning low absolute lung volume rather than low tidal volume). This has been well defined in animal models, but the relevance to humans is not firmly established. Several studies have shown that the injurious effects of mechanical ventilation can be attenuated by the application of PEEP.10,15,20,21 Ventilation with high tidal volume and low or zero PEEP appears to be more damaging than low tidal volume and high PEEP, even though both strategies result in similar high levels of end inspiratory pressure and alveolar distension. Ventilation of isolated lavaged rat lungs with small tidal volumes (5–6 ml/kg) and low or zero PEEP caused lung injury that could be reduced by the application of higher levels of PEEP.22
A number of mechanisms may explain lung injury associated with ventilation at low absolute lung volumes. Cyclic opening and closing (recruitment-derecruitment) of small airways/lung units may lead to increased local shear stress—so called atelectrauma. PEEP effectively splints open the distal airways, maintaining recruitment throughout the ventilatory cycle. The static pressure-volume curve (fig 1) is often used to illustrate the balance between overdistension and recruitment. As airway pressure is increased from functional residual capacity (FRC), an abrupt change in the lung compliance is often evident, particularly in injured or surfactant deficient lungs. This lower inflection point (LIP) may represent the approximate pressure (volume) at which lung units are recruited. The upper inflection point (UIP) at which lung compliance decreases at higher airway pressure was thought to reflect the point at which alveoli are becoming overdistended, and therefore potentially damaged.24 Based on these concepts, an ideal ventilatory strategy would be one in which all the tidal ventilation would take place on the “steep” portion of the pressure-volume curve where the lung is most compliant. The value of PEEP would be sufficient to prevent derecruitment but not so great as to lead to overdistension. High frequency oscillatory ventilation (HFOV) potentially offers the ideal combination of minimum tidal volume while maintaining maximal recruitment (the “open lung”), provided sufficient end expiratory lung volume is maintained.25,26

Pressure-volume curve derived from a patient with ARDS. FRC=functional residual capacity; LIP=lower inflection point; UIP=upper inflection point. Adapted from Matamis et al23 with permission.
Although theoretically sound, the explanation of VILI according to the pressure-volume curve is certainly a gross oversimplification. Recruitment is not complete at the LIP and continues at higher inflating pressures.27,28 Similarly, the UIP does not necessarily reflect the onset of overdistension. Instead, it may represent the point at which recruitment is complete and therefore compliance decreases. Furthermore, inflation may be expanding alveoli without necessarily overdistending them.28 In addition, it is difficult to show that recruitment-derecruitment actually occurs,29 and dynamic ventilation may not follow the pattern of the static pressure-volume curve. Indeed, a recent study using saline lavaged rabbits suggested that ventilation follows the deflation limb of the pressure-volume curve, provided an adequate recruitment manoeuvre is performed.30 Thus, theoretically, a lung could be maintained in a recruited state at lower airway pressures than the inflation limb of the pressure-volume curve would indicate and, conversely, pressures applied on the basis of the inflation limb would cause overdistension. Finally, but of perhaps greatest importance, the damaged lung in the clinical setting is not homogeneously affected, as detailed below. Applied airway pressure that may be ideal to recruit and ventilate some lung units may be inadequate to open the most densely atelectatic regions, and yet simultaneously cause overdistension in the most compliant areas.31,32
Other ventilator parameters
Few studies have addressed the effect of ventilator parameters other than tidal volume, airway pressure, and PEEP in VILI. Increased respiratory frequency may augment lung injury through greater stress cycling, a phenomenon well described in engineering, or through the deactivation of surfactant. Isolated perfused rabbit lungs ventilated with a frequency of 20 breaths/min show greater oedema and perivascular haemorrhage than those ventilated at 3 breaths/min.33 The relevance of these findings to clinical practice is unclear.
Inspired oxygen fraction
Oxidant stress is believed to be an important mechanism in mediating lung injury in a variety of lung diseases including ARDS, and exposure of animals or humans to a high inspired oxygen fraction (Fio2) leads to lung damage, probably through the increased generation of reactive oxygen species.34,35 Current practice in ARDS is to use the lowest Fio2 giving an oxygen saturation of around 90%.
Carbon dioxide
Arterial carbon dioxide tensions (Paco2) reflect minute ventilation. The advent of protective ventilatory strategies in ARDS, with lower tidal volumes and minute ventilation, was accompanied by the acceptance of higher levels of Paco2 (permissive hypercapnia) and mild respiratory acidosis.36 Rather than being harmful, some animal studies suggest that hypercapnia exerts a protective effect in lung injury, although to date there is no clear evidence from clinical studies.37
Patient determinants of VILI
The condition of the ventilated lung is of considerable importance in determining susceptibility to VILI. At one extreme, VILI does not appear to be a clinical problem in patients with normal lungs who can undergo prolonged periods of mechanical ventilation without detrimental effect.38 In these instances the pressures and flows within the lung closely resemble the physiological situation. At the other extreme, the grossly abnormal lungs of patients with ARDS are highly susceptible to VILI, and it may be that in some patients no mechanical ventilation strategy is entirely devoid of detrimental effects.
Ventilating the injured lung
Animal studies using isolated lungs and intact animals have indicated that injured lungs are more susceptible to VILI.39–41 An important factor underlying this predisposition to VILI is the uneven distribution of disease and inflation seen in injured lungs. Based on the diffuse relatively homogeneous distribution of shadowing on a plain chest radiograph, it was thought that the lung was uniformly affected in ARDS. However, CT scanning showed that the posterior dependent portions of the lung are more severely affected (fig 2A), a distribution determined largely by gravity. The greater compliance of less affected areas means that they receive a much greater proportion of the delivered tidal volume.42 This may result in substantial regional overdistension and hence injury.

(A) CT scan of a 25 year old man with ARDS showing the typically heterogeneous distribution of opacification within the lungs, mostly in the posterior dependent regions. (B) CT scan of the same patient 8 months later showing remarkably little abnormality in the posterior regions but with reticular changes anteriorly (large arrows). Reproduced with permission from Desai et al.45
In a recent study, piglets with multifocal pneumonia were ventilated using a tidal volume of 15 ml/kg for 43 hours.43 Approximately 75% of the lung volume was consolidated, so the residual 25% of normally ventilated lung may have received a tidal volume equivalent to 40–50 ml/kg. Histological examination showed emphysema-like lesions in these areas, whereas in consolidated areas the alveoli were “protected” against overdistension, but the bronchioles that remained patent were injured through overdistension and by the forces generated through interdependence and recruitment-derecruitment (fig 3). Lesions similar to those described above have been reported in a necropsy series of patients with ARDS44; furthermore, CT scans of ARDS survivors have shown greatest residual abnormality in the anterior parts of the lung, even though the posterior areas had been most abnormal in the acute phase (fig 2B).45 These changes may be due to injury caused by overdistension.
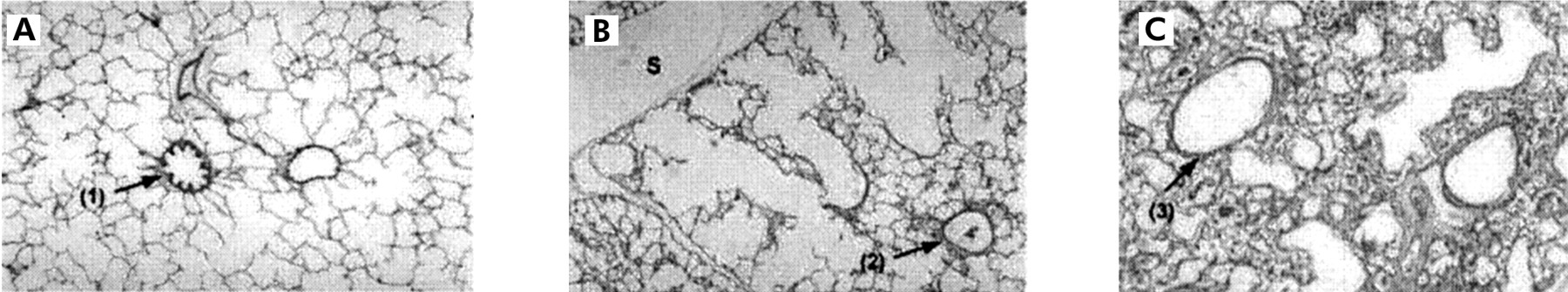
Histological specimens of lung from (A) control piglets and (B) and (C) piglets with experimental pneumonia following ventilation for 2 days. (A) shows normal lung architecture and bronchioles (arrow 1). In piglets with pneumonia there are emphysematous changes in the ventilated regions (B), but bronchioles are of normal size (arrow 2). In consolidated areas (C) there is bronchiolar dilatation (arrow 3). Reproduced with permission from Goldstein et al.43
Other factors that may promote VILI in already injured lungs are surfactant abnormalities and the presence of an activated inflammatory infiltrate which may be further stimulated by mechanical ventilation.
Lung immaturity
The immature lung may be particularly susceptible to VILI.46 The volume of the lung relative to body weight and the number of alveoli are lower in premature infants, making a tidal volume based on weight potentially more injurious. The resilience of the lung tissue is lower, due to less well developed collagen and elastin elements, and surfactant deficiency leads to alveolar instability and favours airway closure. At delivery the fluid filled, surfactant deficient airways of the preterm neonate require high inflation pressures to establish patency, with potential generation of high shear stress. Preterm lambs show evidence of lung injury after only six high volume insufflations.47
MANIFESTATION OF VILI
Pulmonary oedema
Pulmonary oedema is a prominent feature of experimental models of VILI, particularly those using small animals.10,13,48 The high protein content of the oedema fluid suggests that it is, at least in part, due to increased permeability, and experimental studies have implicated changes at both the epithelial and microvascular endothelial barriers. Increased static inflation of fluid filled lung lobes in sheep led to the passage of larger solutes across the epithelium, a finding observed in other models.49,50 Increased microvascular permeability has been shown by the redistribution of 125I-labelled albumin into the extravascular space in mechanically ventilated rats,51 with parallel findings in other species.17,52 In contrast to the clear evidence for increased permeability, relatively little is known about the contribution of hydrostatic pressures to the development of pulmonary oedema in mechanical ventilation. This stems largely from the great difficulty in assessing transmural pressures at the microvascular level. Studies in lambs have indicated that high tidal volume ventilation causes a relatively modest increase in transmural pulmonary vascular pressures.17 Hydrostatic forces may still be important. Firstly, regional differences in lung perfusion and atelectasis may generate far greater filtration forces in some areas53 and, secondly, in the injured lung even small increases in transmural pressure may greatly increase oedema formation.
Morphological changes
The acute structural changes of “injurious” mechanical ventilation have been best defined using small animal models. Under the light microscope the development of oedema is first evident as perivascular cuffing which progresses to florid interstitial oedema and alveolar flooding with continued ventilation at high pressure.10,51 Changes in the endothelium are detectable by electron microscopy within only a few minutes of high airway pressure ventilation of rats, and appear to precede alterations in the epithelium. Some endothelial cells become focally separated from their basement membrane forming intracapillary blebs. Eventually, diffuse alveolar damage is evident; the epithelial surface becomes grossly disrupted in some areas with destruction of type I but sparing of type II cells.15,48
Larger animals have been used to study the longer term effects of mechanical ventilation. In piglets with experimental pneumonia, alveolar damage is seen in the ventilated regions after 2–3 days, with bronchiolar dilatation in the consolidated regions (fig 3).43 An inflammatory reaction also becomes prominent. Ventilation of piglets with high peak airway pressures leads to neutrophil recruitment within 24 hours and fibroproliferative changes after 3–6 days.54 Similarly, conventional ventilation (tidal volume 12 ml/kg) of saline lavaged rabbits led to accumulation of neutrophils in the lungs, as well as severe epithelial damage and hyaline membrane formation. By contrast, animals ventilated with HFOV had minimal injury.55,56
Increased vascular permeability, diffuse alveolar damage, inflammatory cell infiltrates, and later fibroproliferative changes are not specific to VILI. ARDS and other forms of lung injury are associated with identical pathological appearances. The pertinent question for the clinician is the degree to which the changes classically ascribed to ARDS are in fact attributable to VILI?
MECHANISMS OF VILI
The mechanical forces applied through ventilation may have deleterious effects in at least two ways: (1) through physical disruption of the tissues and cells, which depends not only on the magnitude and pattern of the applied stress but also on the resilience of the lung tissue, and (2) through the aberrant activation of cellular mechanisms leading to inappropriate and harmful responses. Both certainly occur, although it is not clear which is more important clinically.
Physical disruption (stress failure)
How forces are generated within the lung
Macklin and Macklin proposed that air leaks are caused by a momentary high pressure gradient between the alveolus and the bronchovascular sheath.4 Air ruptures across the epithelial surface and tracks along the bronchovascular sheath. It may then pass into the interstitium causing pulmonary interstitial emphysema, into the pleural space causing pneumothorax, into the pericardial cavity leading to pneumopericardium, and so on. The endothelium, in close apposition to the epithelial surface, is subject to stress failure due to forces derived both from transpulmonary and intravascular pressures. The extremely thin blood gas barrier (0.2–0.4 μM) permits free gas exchange by diffusion but exposes the capillary to high wall stress, determined by the ratio of the wall tension to thickness.57 In rabbits stress failure occurs at capillary transmural pressures of 52.5 cm H2O (∼40 mm Hg) or greater and the microscopic lesions of endothelial and epithelial disruption are similar to those caused by high volume ventilation.57–59 Importantly, the blood-gas barrier is more prone to stress failure at higher lung volumes, probably due to increased longitudinal forces acting on the blood vessel.60 The forces generated by mechanical ventilation may therefore interact with those due to pulmonary vascular perfusion to magnify lung injury. Isolated rabbit lungs ventilated with a peak static pressure of 30 cm H2O exhibit greater oedema and haemorrhage when perfused with high flow rates corresponding to higher pulmonary artery pressures.61
Interdependence
Adjacent alveoli and terminal bronchioles share common walls so that forces acting on one lung unit are transmitted to those around it. This phenomenon, known as interdependence, is believed to be important in maintaining uniformity of alveolar size and surfactant function.62 Under conditions of uniform expansion all lung units will be subject to a similar transalveolar pressure, approximately equal to the alveolar minus the pleural pressure. However, if the lung is unevenly expanded, such forces may vary considerably. When an alveolus collapses the traction forces exerted on its walls by adjacent expanded lung units increase and these are applied to a smaller area. These forces will promote re-expansion at the expense of greatly increased and potentially harmful stress at the interface between collapsed and expanded lung units. At a transpulmonary pressure of 30 cm H2O it has been calculated that re-expansion pressures could reach 140 cm H2O.62,63 In a necroscopic study of patients who had died with ARDS, expanded cavities and pseudocysts were found particularly around atelectatic areas, suggesting that these forces do indeed play a role in VILI.44
Recruitment-derecruitment
Theoretically, small airways may become occluded by exudate or apposition of their walls.64,65 In either event, the airway pressure required to restore patency greatly exceeds that in an unoccluded passage. The resulting shear stress may damage the airways, particularly if the cycle is repeated with each breath (∼20 000 times per day). The pressure required to reopen an occluded airway is inversely proportional to its diameter,66 which is consistent with the observation that small airway damage in isolated lungs ventilated at zero PEEP occurs more distally as PEEP is applied.22
Airway collapse is favoured by surfactant deficiency or conditions in which the interstitial support of the airways is weakened or underdeveloped.66,67 Conversely, recruitment-derecruitment may not occur at all in normal lungs, which tolerate periods of negative end expiratory pressure without evident harm.68
Importance of surfactant
Surfactant plays a role in VILI in two related ways; firstly, surfactant dysfunction or deficiency appears to amplify the injurious effects of mechanical ventilation and, secondly, ventilation itself can impair surfactant function thereby favouring further lung damage.
Abnormalities of the surfactant system may contribute to injury in the mature lung as in preterm infants. Surfactant isolated from patients with ARDS and experimental models of pneumonia is functionally impaired.69,70 This is associated with a decrease in the functionally active large aggregate (LA) component of the surfactant pool relative to the less active small aggregate (SA) component. Although stretch of type 2 cells in vitro and a modest degree of hyperventilation stimulate surfactant production,71 more injurious ventilatory strategies of high tidal volume and low PEEP lead to a reduced pool of functional surfactant and a decreased LA:SA ratio, particularly in injured lungs.72 Large cyclical alterations in the alveolar surface area and the presence of serum proteins in the airspace may be responsible for these changes.
Lungs can be rendered experimentally deficient in surfactant by saline lavage or detergent aerosolisation, and they may behave in a manner similar to those of premature infants. Strategies that maintain lung recruitment, such as HFOV (with recruitment manoeuvres) and conventional mechanical ventilation at high levels of PEEP, appear to be particularly effective in minimising lung injury in these models.73–76 Surfactant abnormalities may contribute to VILI in several ways relating to the increase in surface tension:
Alveoli and airways are more prone to collapse with generation of shear stress as they are reopened.
The uneven expansion of lung units increases regional stress forces through interdependence.
The transvascular filtration pressure is increased, promoting oedema formation.77
In addition, surfactant is thought to have important immunoregulatory functions78 which may become impaired through mechanical ventilation.
From the preceding discussion it is logical to propose that increasing the pool of functioning surfactant might lessen lung injury. Surfactant therapy reduces mortality in the neonatal respiratory distress syndrome and may decrease lung injury.79 In ARDS a role for surfactant supplementation is not established,80 partly because of difficulties in delivering adequate amounts of active surfactant to damaged and collapsed lung regions.
Activation of aberrant cellular pathways
Physical forces such as stretch play an important role in physiological processes. In fetal life breathing is essential for lung development and in the mature lung ventilation stimulates surfactant production by type II pneumocytes.71 Central to this is the concept of mechanotransduction, whereby physical forces are detected by cells and converted into biochemical signals. There is now good evidence that signalling events activated by injurious ventilation play a role in VILI.81,82
The increase in lung vascular permeability induced in isolated perfused rat lungs by high airway pressure ventilation can be blocked by gadolinium in the perfusate.83 Gadolinium probably exerts this effect through its inhibition of stretch activated cation channels. This indicates that the oedema seen in injurious ventilation is, at least in part, due to the activation of specific cellular processes rather than simply being a reflection of physical disruption of the alveolar-capillary barrier (the “stretched pore” phenomenon).
Considerable attention has focused recently on the release of inflammatory mediators from lung tissue exposed to mechanical forces. A number of studies involving isolated lungs or intact lung injured animals of different species have shown that injurious ventilatory strategies are associated with the release of a variety of proinflammatory mediators, including thromboxane B2, platelet activating factor, and several cytokines.84–88 This humoral inflammatory response can precede overt histological damage and appears to be due to stretch activation of specific pathways, in addition to an inflammatory reaction to non-specific injury.82,88 The importance of these mediators in causing lung injury is unknown, and it is conceivable that they exert a beneficial effect in the injured lung.89 However, studies using rabbit models of VILI have shown that lung damage can be attenuated by administration of anti-tumour necrosis factor (TNF)-α antibodies or interleukin (IL)-1 receptor antagonists, suggesting that these cytokines exert a deleterious effect.90,91 In the preterm lung cytokines generated by mechanical ventilation may interfere with lung development.46 The term biotrauma has been coined to describe this potentially injurious inflammatory response to physical stress.
Mediators generated in response to injurious ventilation do not remain compartmentalised within the lung. Experiments with perfused mouse lungs and with lung injured rats in vivo have indicated that injurious ventilation leads to increased cytokine levels in the systemic circulation, and recent studies suggest that the same applies in the clinical setting.86,87,92,93 This has led to the hypothesis that mechanical ventilation can fuel the systemic inflammatory response commonly seen in ARDS and contribute to the development of multiple system organ failure (MSOF).94
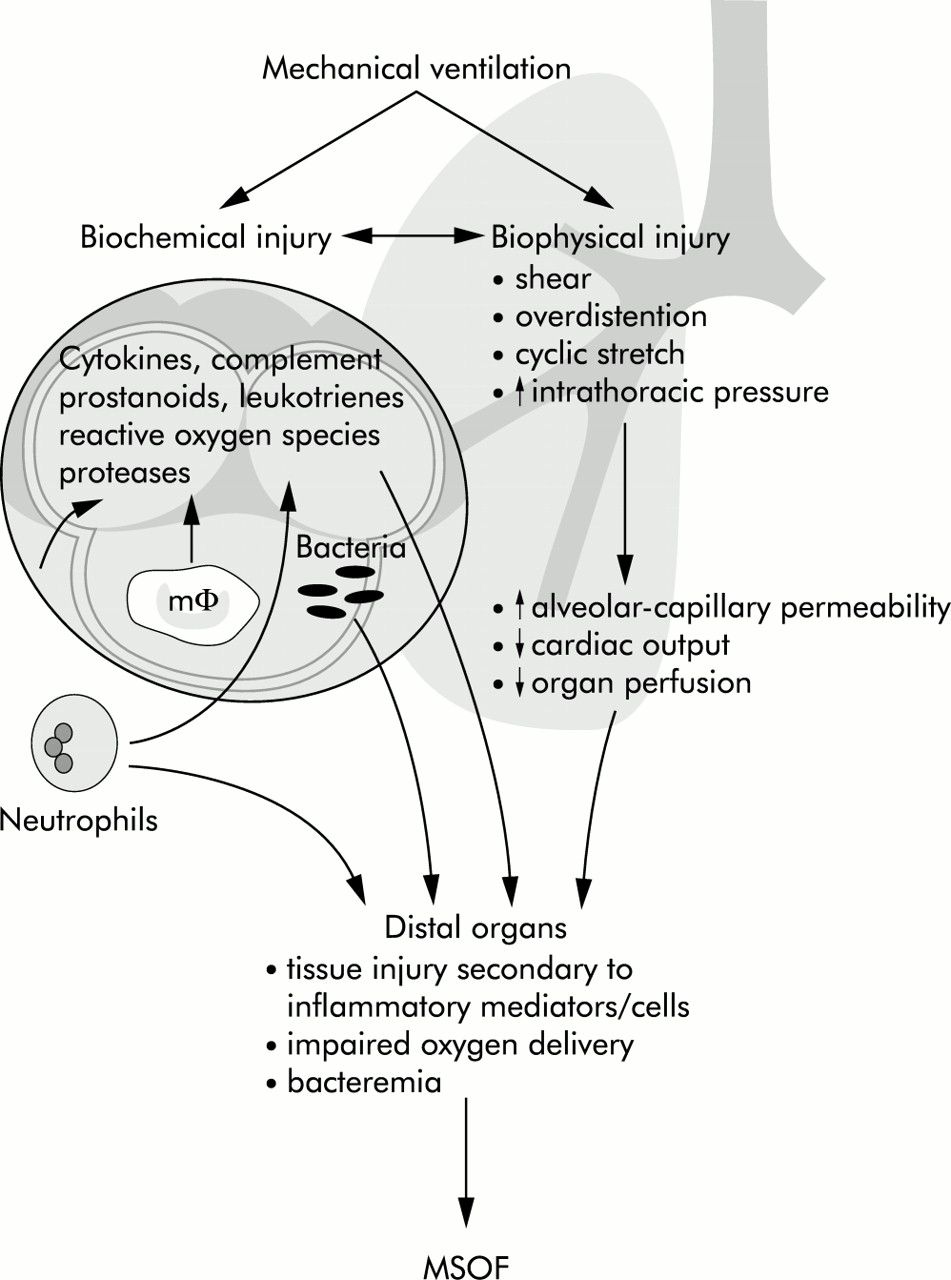
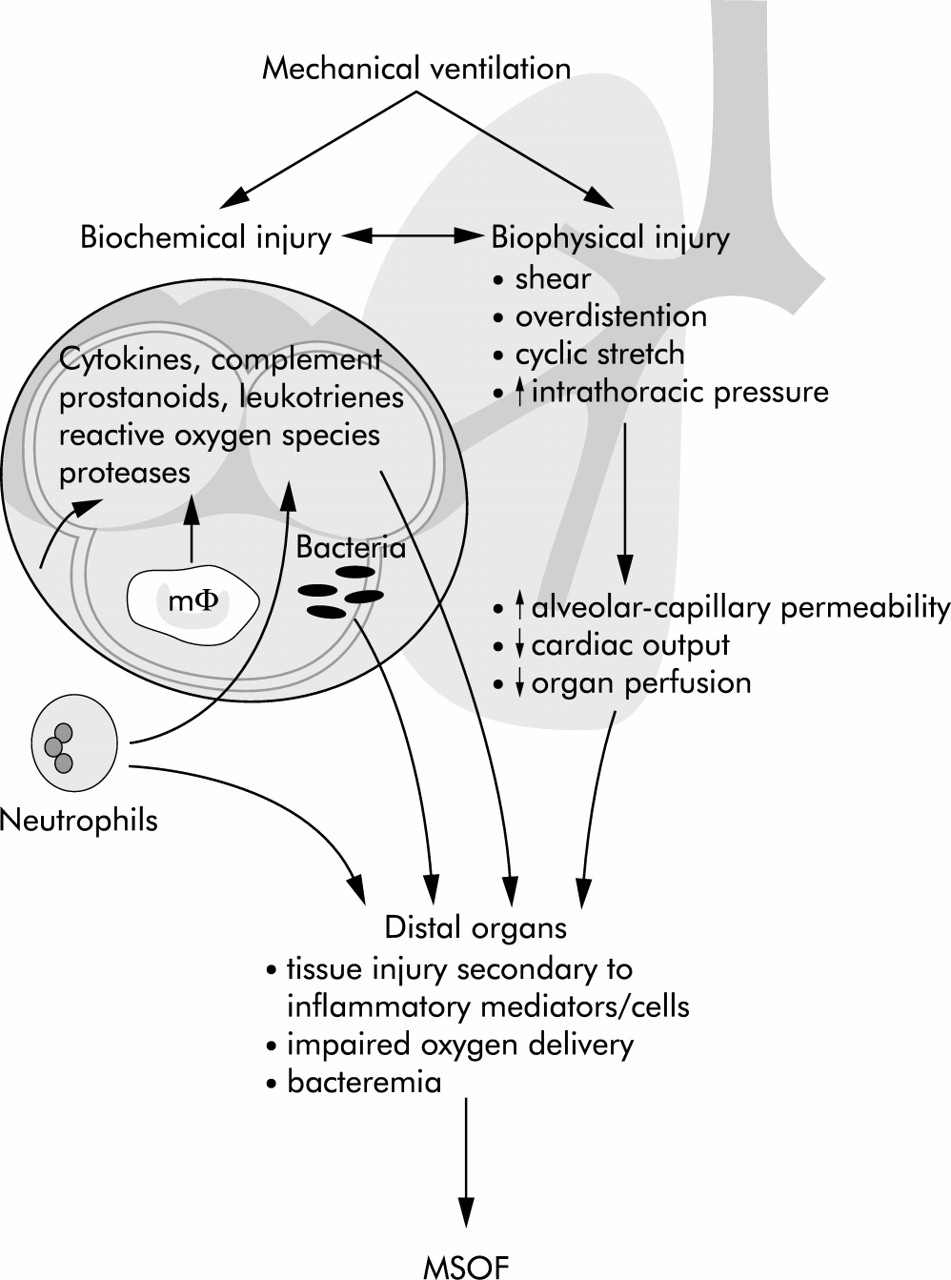
Ventilation may also influence the systemic inflammatory response through translocation of bacteria or their products from the air spaces into the circulation. In dogs and rats bacteraemia is more likely to develop when lungs that have been inoculated with bacteria are ventilated with high tidal volume/zero PEEP compared with less injurious strategies.95,96 An analogous effect has been observed in ventilated rabbits following intratracheal administration of lipopolysaccharide (LPS). Injurious ventilation resulted in much higher levels of circulating LPS accompanied by a rise in TNF-α.97 The possible ways in which ventilation impacts on systemic inflammation and distal organs are summarised in fig 4.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mechanisms by which mechanical ventilation might contribute to multiple system organ failure (MSOF). Reproduced with permission from Slutsky and Tremblay.94
CLINICAL CONSEQUENCES OF VILI
Despite the difficulties in distinguishing the effects of mechanical ventilation from those of the underlying condition, there are now clear data showing the clinical impact of VILI in two conditions—neonatal respiratory distress syndrome and ARDS.
Neonatal chronic lung disease
Most cases of neonatal chronic lung disease (CLD), also known as bronchopulmonary dysplasia, occur in the aftermath of neonatal respiratory distress syndrome. Hyperoxia and mechanical ventilation have been implicated in its aetiology.98 Evidence for the role of mechanical ventilation in this respect includes the observation that neonatal intensive care units with high rates of intubation and ventilation also have high rates of CLD without improvements in mortality or other morbidity.99,100 At two centres with very different rates of mechanical ventilation of low birth weight infants (75% v 29%) and prevalence of CLD (22% v 4%) multivariate regression analysis indicated that the development of CLD was strongly associated with the initiation of mechanical ventilation.100 Several trials have addressed the use of different ventilatory strategies, particularly HFOV, in neonatal respiratory distress. Despite its theoretical advantages over conventional ventilation in terms of reducing lung injury, the role of HFOV in neonatal respiratory distress remains controversial.26,101
ARDS
The magnitude of the clinical burden of VILI was shown by the recent ARDSnet trial in which 861 patients with ARDS were randomised to receive either a “traditional” tidal volume (12 ml/kg predicted body weight) or a low tidal volume strategy (6 ml/kg).2 Mortality was 39.8% in the traditional group and 31% in the low volume group. In other words, at least 8.8% of the absolute mortality from ARDS is attributable to VILI. A considerable amount of attention is currently directed at the potential clinical benefits of improving and maintaining lung recruitment, based on the theories outlined above, using higher levels of PEEP or HFOV.
It is interesting to speculate on precisely how VILI increases mortality. The injury to the lung described in experimental models probably occurs in humans, to a greater or lesser degree. However, most deaths in ARDS are from MSOF rather than respiratory failure.102 It is therefore likely that mechanical ventilation can influence the development of MSOF, possibly through the release of proinflammatory mediators, as described above. Two recent clinical studies add weight to this hypothesis. Ranieri and coworkers examined the effect of two ventilatory strategies on cytokine levels in ARDS. Forty four patients were randomised either to a “protective” strategy, in which the PEEP and tidal volume were set such that tidal ventilation occurred exclusively between the lower and upper inflection points of the pressure volume curve (fig 1), or a “control” strategy in which the tidal volume was set to obtain normal values of arterial CO2 and the PEEP set to produce the greatest improvement in arterial oxygen saturation without worsening haemodynamics. The protective group had significantly lower levels of plasma and bronchoalveolar lavage cytokines and significantly less organ failure.93,103 Along similar lines, the ARDSnet study found that plasma levels of IL-6 fell significantly more in patients ventilated with the lower than the traditional tidal volume.2 It therefore seems likely that mechanical ventilation in ARDS can promote systemic inflammation and multiorgan failure. Crucially, however, it is not known which factor(s) are responsible for mediating this detrimental effect and how they exert a toxic effect on distal organs.
In addition to increasing mortality in ARDS, VILI may contribute to the persistent lung function abnormalities (principally a restrictive defect with abnormal transfer factor) seen in a minority of survivors.104 However, no studies to date have shown that protective ventilatory strategies in ARDS are associated with improved long term lung function.105
CONCLUSIONS
Extensive research over the past 30 years has identified key determinants of VILI. From this, practice has been modified in an attempt to minimise lung damage. In the case of ARDS, ventilation with lower tidal volumes has been shown to reduce mortality.
One of the most exciting developments has been the realisation that VILI may be caused not only by the mechanical disruption of lung tissue, but also by the inappropriate activation of cellular pathways. Such mechanisms may contribute to non-pulmonary organ damage. Future treatments to minimise the impact of VILI may target these mechanisms at the molecular level, in addition to developing less injurious ventilation strategies.
Acknowledgments
TW is supported by the Scadding-Morriston-Davies Trust. AS is supported by the Canadian Institutes of Health Research (grant # 8558).