Article Text

Abstract
Background: A special regulatory role for prostaglandin E2 has been postulated in aspirin-induced asthma. A study was undertaken to investigate the effects of aspirin on the systemic production of prostaglandin E2 and cysteinyl leucotrienes in patients with asthma.
Methods: The urinary concentrations were determined of two main prostaglandin E2 metabolites (13,14-dihydro-15keto-PGE2 using a commercial enzyme immunoassay and 9,15-dioxo-11α-hydroxy-2,3,4,5-tetranor-prostane-1,20-dioic acid by gas chromatography/mass spectrometry) and leucotriene E4 using an immunoassay. Determinations were performed at baseline and following oral aspirin and celecoxib challenges in two well-defined asthma phenotypes: aspirin-sensitive and aspirin-tolerant patients.
Results: Aspirin precipitated bronchial reactions in all aspirin-sensitive patients but in none of the aspirin-tolerant patients. Celecoxib 400 mg was well tolerated by all patients except for one with aspirin-induced asthma. At baseline, the mean levels of prostaglandin E2 metabolites did not differ between the groups. Following different aspirin provocation doses, the mean levels of the two main prostaglandin E2 metabolites were decreased in the aspirin-tolerant group but remained unchanged in the aspirin-sensitive group. The dose of aspirin had no effect on the magnitude of the response on the prostaglandin E2 metabolites and its duration. In both groups, urinary prostaglandin E2 metabolites decreased following celecoxib challenge. No correlation was found between prostaglandin E2 metabolites and leucotriene E4.
Conclusions: Aspirin-precipitated asthmatic attacks are not associated with changes in the systemic production of prostaglandin E2. In contrast, the systemic production of prostaglandin E2 becomes depressed by aspirin in non-sensitive patients. This different response might indicate COX-1-dependent prostaglandin E2 control of inflammatory cells in aspirin-induced asthma. Thus, PGE2 is released during the clinical reactions to aspirin through an alternative COX-2 pathway. The clinical implications of this finding are in line with current observations of good tolerance of the selective COX-2 inhibitors in aspirin-sensitive patients.
Statistics from Altmetric.com
Prostaglandin E2 (PGE2) is a bioactive compound formed by actions of cyclooxygenase (COX) and specific PGE synthases.1 In human airways PGE2 is produced by many cells including epithelium, smooth muscle, alveolar cells, macrophages, phagocytes and lymphocytes.1 2 In vitro, PGE2 relaxes smooth muscle and displays a number of inhibitory effects on mast cell degranulation, synthesis of leucotriene B4, activation of granulocytes and T cells.3–5 PGE2 elicits a large number of biological effects acting through four receptors: EP1, EP2, EP3 and EP4.6–8 The response of target cells to PGE2 varies according to the spectrum of receptors they express.
PGE2 might be of special importance in aspirin-induced asthma. This is a distinct clinical syndrome affecting 5–10% of adults with asthma.9 10 Asthma attacks triggered by aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs) are associated with inhibition of COX11 12 (specifically COX-1 but not COX-2) and are characterised by overproduction of cysteinyl-leucotrienes (cys-LTs).13–15 Inhaled PGE2 protects against both aspirin-precipitated attacks of asthma and the massive release of urinary LTE4.16 17 The protection does not seem to be achieved through relaxant effects on bronchial smooth muscle, but rather by suppression of the mediators released from the proinflammatory cells. The incriminated cells could be mast cells or eosinophils. PGE2 greatly inhibits oxygen bursts in granulocytes, including eosinophils,4 and slows down the biosynthesis of cys-LTs in peripheral blood mononuclear cells.18
Because of the rapid metabolism of PGE during sampling and isolation of plasma,1 measurement of stable metabolites in urine rather than the parent compound is the most efficacious method of assessing their endogenous production.19 Moreover, human kidneys are abundant source of prostanoids, including PGE2. Local biosynthesis of PGE2, dependent on both constitutive COX-1 and inducible COX-2 in the kidneys, varies with age and salt intake. It is also affected by several drugs including angiotensin converting enzyme inhibitors.20 PGE2 produced in the kidneys is excreted as the parent compound and does not affect the pool of metabolites produced by systemic inactivation of PGE2.21 The main urinary metabolites of PGE2 levels are believed to reflect global PGE2 production. The predominant pathway of PGE2 metabolism has been shown to involve transformation of PGE2 by the 15-OH PGDH enzyme to 15-keto-PGE2. Subsequently, 15-keto-PGE2 is rapidly converted to its main metabolite of PGE2 (13,14-dihydro-15keto-PGE2; PGE2-M) by the enzyme 15-keto-prostaglandin-Δ13 reductase.22 Oxidation of 13,14-dihydro-15keto-PGE2 leads to formation of a major metabolite of PGE2 (9,15-dioxo-11α-hydroxy-2,3,4,5-tetranor-prostan-1,20-dioic acid; tetranor-PGE-M). Tetranor-PGE-M is a stable urinary metabolite of PGE1 and PGE2 and is used as a urinary marker of PGE2 biosynthesis.23 24
We studied the urinary excretion of two major PGE2 metabolites, reflecting the systemic production of this prostaglandin. The studies were carried out in patients with aspirin-sensitive and aspirin-tolerant asthma, both at baseline and after challenge with aspirin and celecoxib. We expected to be able to identify aspirin intolerance by a decrease in systemic PGE2 production following aspirin challenge. We also looked at the possible relationship between release of PGE2 metabolites and that of cys-LTs. To our knowledge, we are the first to perform such investigations.
METHODS
Subjects
The study population consisted of 19 patients with aspirin-induced asthma (AIA) and 21 patients with aspirin-tolerant asthma (ATA). The characteristics of the study patients are shown in table 1.
The diagnosis of aspirin intolerance was confirmed by oral aspirin provocation tests performed during the 24 months preceding the study. All patients with ATA occasionally used aspirin without any adverse reactions. The patients had stable asthma and their baseline forced expiratory volume in 1 s (FEV1) was >70% of the predicted value on the study day. None had experienced an exacerbation or a respiratory tract infection in the 6 weeks preceding the study. The subjects were instructed to withhold medications that decrease bronchial responsiveness prior to aspirin/celecoxib challenge. Short-acting β2 agonists were not used 8 h before the challenge. Long-acting β2 agonists and theophylline were withdrawn for 24 h. Short-acting antihistamines and cromones were stopped 5 days before the challenge. Inhaled steroids were allowed at a dose of ⩽2000 μg budesonide per day. None of the patients was treated with systemic corticosteroids or leucotriene modifying drugs.
Basal urinary levels of PGE2-M, tetranor-PGE-M and LTE4 were measured in 30 healthy subjects. The controls tolerated NSAIDs well and had no history of adverse reactions to aspirin and other aspirin-like drugs.
The patients gave informed consent and the study was approved by the university ethics committee.
Study design
The study consisted of two phases. In the first phase, at 1-week intervals, the patients underwent aspirin and celecoxib testing. The single-blind placebo-controlled oral challenge test with aspirin was carried out on two consecutive days. On the first day four capsules of placebo were administered every 1.5 h. On the second day the patients were challenged with increasing doses of 27, 44, 117, 312 mg aspirin at 1.5 h intervals up to the cumulative dose of 500 mg.25 On the eighth day a single dose of 400 mg celecoxib was administered.
In the second phase of the study, following a 2-week run-in period, the same patients with ATA again underwent provocation tests with aspirin at a cumulative dose of 188 mg (ie, the dose which precipitated asthma attacks in nine patients with AIA). The purpose of the second phase was to exclude a dose dependence of the results obtained in the first phase in the ATA group. In the first phase, all patients with ATA were given aspirin in a dose of 500 mg while provocation doses of aspirin among patients with AIA differed. A single-blind placebo-controlled oral challenge test with aspirin was carried out on two consecutive days. On the first day three capsules of placebo were administered every 1.5 h. On the second day the patients were challenged with increasing doses of 27, 44 and 117 mg aspirin at 1.5 h intervals up to the cumulative dose of 188 mg.
Placebo, aspirin and celecoxib had an identical appearance. The challenge procedure with aspirin and/or celecoxib was interrupted if a bronchospastic reaction occurred (FEV1 fell ⩾20%) or if the maximum cumulative dose of aspirin and a single dose of celecoxib was reached. The cumulative dose of aspirin causing a 20% fall in FEV1 was calculated and recorded as the provocation dose of aspirin (PD20).
FEV1 and extrabronchial symptoms were recorded at baseline, before the challenge tests and then every 30 min until 6 h after the last dose of aspirin and celecoxib.
In patients with a positive aspirin challenge (AIA), urine samples were collected for measurement of PGE2-M, tetranor-PGE-M and LTE4 estimations performed at baseline, at the time of appearance of the bronchial symptoms (time 0) and 2 and 4 h later. In patients with ATA in whom the aspirin challenge was negative, urine samples were collected at baseline, 1.5 h after the last aspirin dose (ie, when the cumulative doses of 500 mg (first phase) and 188 mg (second phase) were reached (time 0)), and then 2 and 4 h later.
In case of a single dose of celecoxib, urine samples were collected in the same manner as for the aspirin provocation challenge in patients with ATA and AIA.
Lung function tests
Pulmonary function tests were performed on a flow-integrating computerised pneumotachograph (Pneumoscreen; E Jaeger, Germany).
Urinary levels of PGE2-M, tetranor-PGE-M and LTE4
Urinary levels of PGE2-M (Cayman Chemical, Prostaglandin E metabolite EIA Kit) and LTE4 (Cayman Chemical, Ann Arbor, Michigan, USA) were measured in unpurified urine samples by direct enzyme immunoassay.26 Measurements were made at the same time, in duplicates, using the same batch of the reagents. The results were expressed as pg/mg creatinine. The urinary concentration of tetranor-PGE-M was measured by gas chromatography/mass spectrometry27 28 (see methods in the online data supplement).
Statistical analysis
Summary statistics were expressed as mean (M), standard deviation (SD), median (Me) and 25% and 75% percentiles. General Linear Model (GLM) including repeated measures analysis of variance, which takes into account the fact that the outcome measurements are repeated over time within subjects, was used for multiple comparisons. Logarithmic transformation was used when needed as variance stabilising transformation. To describe better the changes in time for log-transformed data, 95% confidence intervals (CI) in fold difference units29 were constructed.
Correlation between variables was estimated with the Spearman rank order correlations. A p value of ⩽0.05 was considered statistically significant.
RESULTS
Clinical reactions
There was no statistical difference in clinical characteristics between the patients with AIA (positive aspirin challenge test) and those with ATA (negative aspirin challenge test), table 1. None of the patients developed symptoms after administration of placebo. In the AIA group, bronchial reactions developed after 27 mg in one subject, after 44 mg in one subject, after 117 mg in nine subjects and after 500 mg in six. The mean cumulative dose of aspirin was 188 mg. One patient developed asthma and anaphylactic shock after receiving aspirin at a dose of 177 mg. In another patient, abdominal pain accompanied by a transient increase in urinary and serum amylase (6718 U/l (normal range 32–640 U/l) and 2031 U/ml (normal range 30–110 U/ml), respectively) and serum lipase (615 U/l (normal range 23–300 U/l)) was recorded.
None of the patients with ATA developed any clinical symptoms following aspirin or celecoxib challenges. In one of 17 patients with AIA the celecoxib challenge produced dyspnoea and FEV1 fell by 21%. This was the same patient who developed shock after aspirin administration. The case has been described elsewhere.30 The remaining 16 patients in the AIA group tolerated celecoxib very well.
All the symptoms were relieved by short-acting β2 agonists. Systemic corticosteroids were required in three cases only. Subcutaneous adrenaline was administered to the patient who developed shock.
Urinary PGE2-M levels
Phase I
At baseline, urinary levels of PGE2-M did not differ significantly between the study groups and healthy control subjects (table 2, p = 0.52). After placebo administration, no significant differences in urinary PGE2-M levels were found between the study groups (p = 0.58, ANOVA). In patients with AIA, urinary PGE2-M levels increased 2 h following placebo administration compared with baseline values (p = 0.01; 95% CI 1.030 to 1.722 baseline; fig 1A).
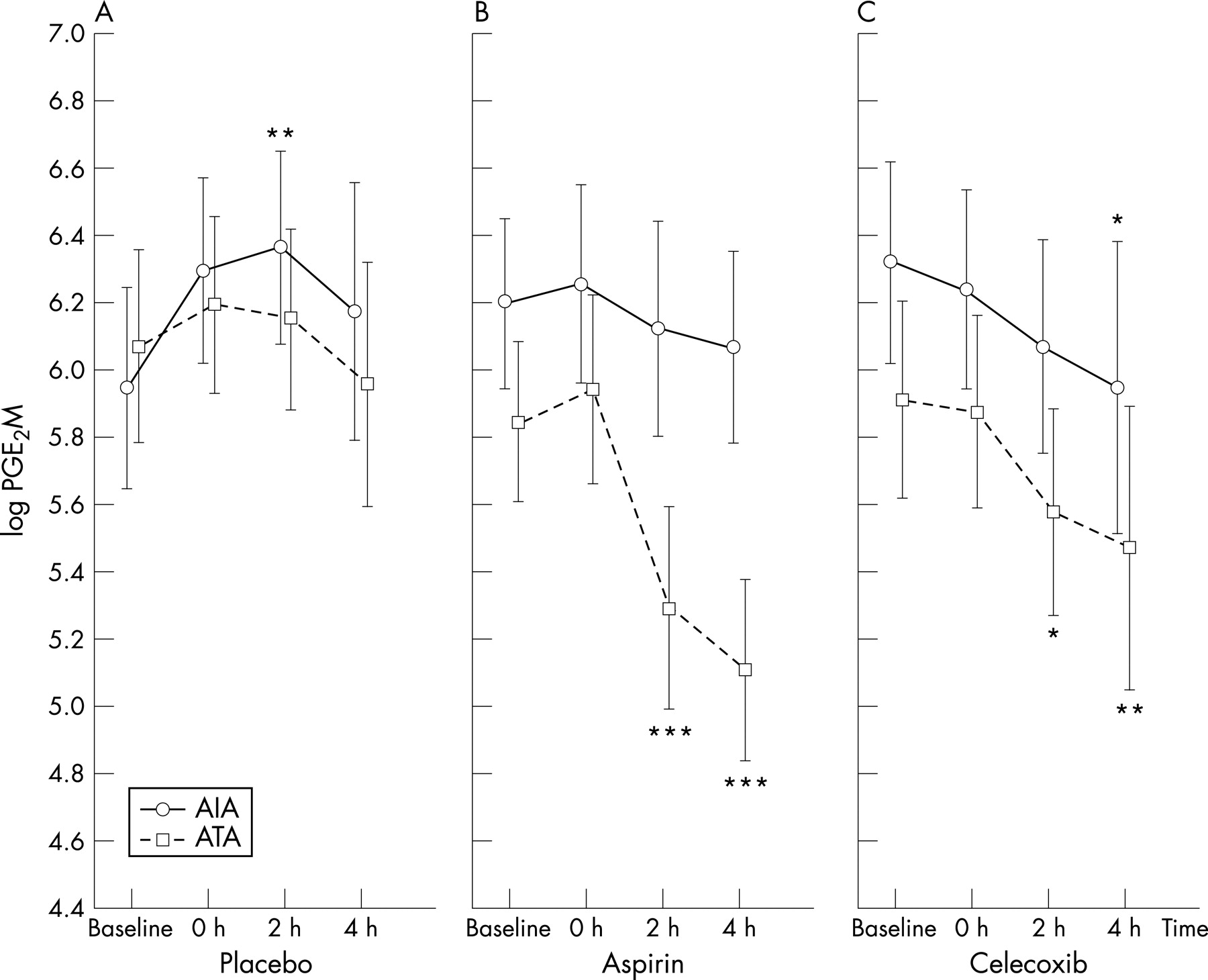
PGE2-M values on the day of aspirin challenge differed significantly between the AIA and ATA groups (p<0.001, ANOVA). The levels did not change at any time during the observation period in patients with AIA but, in the ATA group, urinary PGE2-M concentrations were decreased 2 h (p<0.001; 95% CI 0.417 to 0.688 baseline) and 4 h (p<0.001; 95% CI 0.339 to 0.559 baseline) following aspirin challenge tests compared with baseline values. The lowest level was reached 4 h after aspirin administration (fig 1B).
Following celecoxib challenge, urinary concentrations of PGE2-M were significantly higher in patients with AIA (p = 0.04, ANOVA). In the AIA and ATA study groups urinary PGE2-M levels decreased at 2 h (p = 0.08 and p = 0.05; 95% CI 0.557 to 0.918 baseline, respectively) and 4 h (p = 0.04; 95% CI 0.532 to 0.889 baseline and p = 0.007; 95% CI 0.521 to 0.857 baseline, respectively) following celecoxib challenge tests compared with baseline values (fig 1C). The dose of aspirin had no effect on the magnitude of the response of PGE2-M and its duration.
At baseline and 4 h following aspirin challenge, urinary PGE2-M levels were increased only in patients with AIA with the highest levels of total serum IgE (Spearman r = 0.6; p = 0.03). Baseline concentrations of PGE2-M in the urine of healthy controls were 1.59-fold (59%) greater in men than in women (p = 0.01). In contrast, there was no relationship between gender and urinary PGE2-M levels at baseline or following any of the challenges in the patient groups studied (p = 0.18, ANOVA).
Phase II
At baseline, urinary levels of PGE2-M did not differ significantly between placebo and aspirin days in patients with ATA (p = 0.38). After placebo administration, no significant differences in urinary PGE2-M levels were found. Urinary PGE2-M levels decreased 2 h (p = 0.005; 95% CI 0.488 to 0.737 baseline) and 4 h (p<0.001; 95% CI 0.399 to 0.603 baseline) following a cumulative dose of 188 mg aspirin compared with baseline values in patients with ATA (fig 2A).

Urinary 9,15-dioxo-11α-hydroxy-2,3,4,5-tetranor-prostane-1,20-dioic acid (tetranor-PGE-M)
Phase I
At baseline, urinary levels of tetranor-PGE-M did not differ significantly between both study groups and healthy control subjects (table 2, p = 0.94, ANOVA). Following placebo administration, no significant differences in urinary tetranor-PGE-M levels were found between patients with AIA and those with ATA (p = 0.55, ANOVA). The levels did not change at any time during the observation period (p = 0.99, ANOVA, fig 3A).
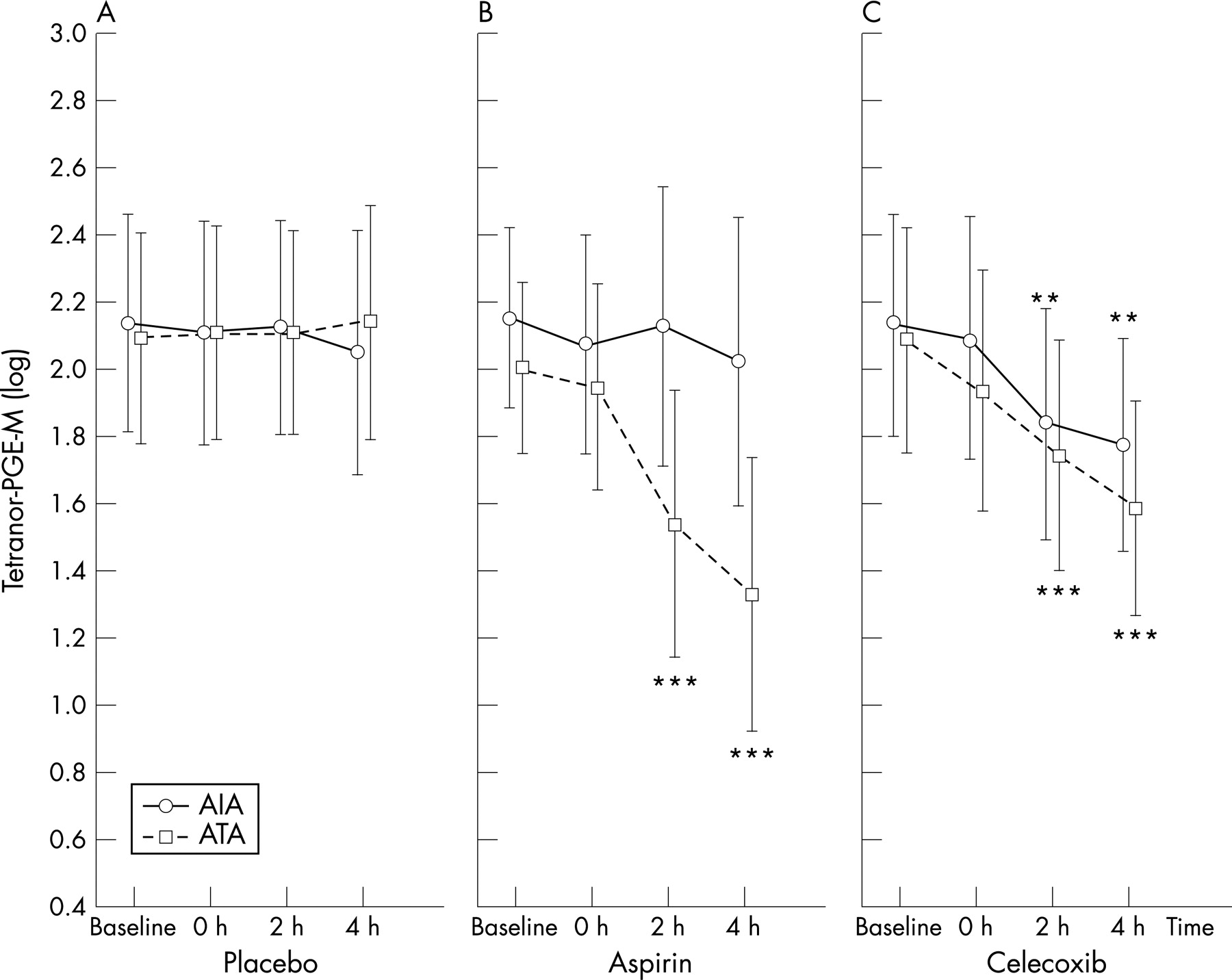
Tetranor-PGE-M levels following aspirin challenge did not change at any time of the observation period in patients with AIA (p = 0.23, ANOVA) but, in patients with ATA, the levels decreased significantly (p<0.001, ANOVA). In the ATA group, urinary concentrations of tetranor-PGE-M decreased at 2 h (p = 0.001; 95% CI 0.489 to 0.748 baseline) and 4 h (p<0.001; 95% CI 0.391 to 0.615 baseline) following aspirin challenge tests compared with baseline values (fig 3B).
Following celecoxib challenge, no significant differences were found in urinary tetranor-PGE-M levels between patients with AIA and those with ATA (p = 0.58, ANOVA). Urinary tetranor-PGE-M levels in the AIA and ATA groups decreased at 2 h (p<0.001; 95% CI 0.586 to 0.933 baseline and p<0.001; 95% CI 0.565 to 0.888 baseline, respectively) and 4 h (p = 0.009; 95% CI 0.352 to 0.878 baseline and p<0.001; 95% CI 0.483 to 0.759 baseline, respectively) following celecoxib challenge compared with baseline values (fig 3C).
The dose of aspirin had no effect on the magnitude of the response of tetranor-PGE-M and its duration (p = 0.32, ANCOVA). The concentration of urinary tetranor-PGE-M in individual patients with AIA did not depend on the cumulative dose of aspirin which caused bronchospasm.
Baseline urine concentrations of tetranor-PGE-M in healthy controls were 1.39-fold (39%) greater in men than in women (p = 0.03). In contrast, there was no relationship between gender and urinary tetranor-PGE-M levels at baseline or following any of the challenges in the patient groups studied (p = 0.26, ANOVA).
Phase II
At baseline, urinary levels of tetranor-PGE-M did not differ significantly between placebo and aspirin days in patients with ATA (p = 0.51). No significant differences in urinary tetranor-PGE-M levels were found after administration of placebo. Urinary tetranor-PGE-M levels decreased 2 h (p<0.001; 95% CI 0.558 to 0.962 baseline) and 4 h (p<0.001; 95% CI 0.466 to 0.887 baseline) following a cumulative dose of 188 mg aspirin compared with baseline values in patients with ATA (fig 2B).
Urinary LTE4
Baseline urinary LTE4 excretion (table 2) was higher in patients with AIA than in those with ATA (p<0.001, ANOVA) or healthy control subjects. In the AIA group a gradual decrease in the urinary LTE4 level was seen following placebo administration reaching at time 0 (p<0.001; 95% CI 0.317 to 0.751 baseline) which continued to fall at 2 h (p<0.001; 95% CI 0.237 to 0.561 baseline) and 4 h (p<0.001; 95% CI 0.166 to 0.394 baseline).
Urinary LTE4 levels increased at 2 h (p = 0.001; 95% CI 2.087 to 4.948 baseline) and 4 h (p = 0.003; 95% CI 1.931 to 4.579 baseline) following aspirin challenge tests compared with baseline values only in the patients with a positive aspirin challenge.
In patients with AIA, following celecoxib challenge urinary LTE4 levels decreased at time 0 (p = 0.02; 95% CI 0.321 to 0.760 baseline) and remained stable 2 h (95% CI 0.320 to 0.762 baseline) and 4 h later (95% CI 0.359 to 0.851 baseline). There were no changes in urinary LTE4 levels after celecoxib challenge in patients with ATA compared with baseline values (fig 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
There was a positive correlation between the urinary LTE4 levels (at baseline and following aspirin challenge) and the blood eosinophil count only in patients with AIA (Spearman r = 0.78; p<0.001).
Analysis of correlation between urinary LTE4 and PGE2-M levels
Following aspirin challenge, no correlation was found between urinary LTE4 and PGE2-M levels in patients with AIA. There was a positive correlation between urinary LTE4 levels and the levels of PGE2-M following aspirin challenge (at 4 h) only in patients with ATA (Spearman r = 0.63; p = 0.006). No correlation was found in either study group following placebo and celecoxib challenges.
Analysis of correlation between urinary LTE4 and tetranor-PGE-M levels
Following placebo, aspirin and celecoxib challenges, no correlation was found between urinary LTE4 and tetranor-PGE-M levels in patients with AIA (p = 0.99, p = 0.27, p = 0.58, respectively) or ATA (p = 0.37, p = 97, p = 0.52, respectively).
DISCUSSION
In this study we measured the two main metabolites of PGE2 found in the urine: PGE2-M by enzyme immunoassay and tetranor-PGE-M by gas chromatography/mass spectrometry. The latter method discriminated between genuine PGE2 metabolites and the PGE1 end products depending on diet. It has been reported that the mass spectrometry measurement of PGE2 metabolites in urine is highly accurate and sensitive.31
Our results showed a similar baseline urinary level of PGE2-M and tetranor-PGE-M in patients with AIA, those with ATA and healthy controls. In healthy subjects, as has been reported previously,32 the level of both PGE2 metabolites was significantly higher in men than in women. However, we did not observe these differences in patients with asthma. Aspirin challenge in patients with AIA had no effect on PGE2-M and tetranor-PGE-M levels at any time during the observation period compared with baseline values. In contrast, in patients with ATA, urinary levels of both PGE2 metabolites became significantly depressed following two aspirin challenges of either 500 mg or 188 mg. There is a tentative explanation for a paradoxical finding of urinary PGE2 metabolites not changing during the positive aspirin challenge in patients with AIA. These metabolites decreased following comparable doses of aspirin in other subjects not sensitive to aspirin. Constitutively expressed COX-1, by its main product PGE2, controls activation of inflammatory cells. Upon inhibition with aspirin, it is the second source of PGE2 (ie, COX-2) which takes part and may contribute to urinary metabolites. This unmasking effect of aspirin was present only in patients with AIA and, as expected, celecoxib did not discriminate for aspirin sensitivity when urinary metabolites of PGE2 were measured.
It has been postulated that COX-1 inhibition by aspirin, resulting in reduced PGE2 production, provides the mechanism that triggers attacks of asthma.33 This hypothesis has been supported by the following data:
Inhalation of exogenous PGE2 prevents bronchoconstriction provoked by aspirin and inhibits urinary excretion of cys-LTs.16 17 33
Epithelial cells from surgically removed nasal polyps of patients with AIA produce less PGE2 than the same cells from patients with ATA.34
Peripheral blood cells from patients with AIA release less PGE2 at baseline than those from healthy controls.35
PGE2 production by bronchial fibroblasts in patients with AIA is lower than in patients with ATA.36
Lower expression of COX-2 in patients with AIA results in deficient production of PGE2 in nasal polyps, thus contributing to an imbalance between eicosanoids.37 38
Inflammatory cells infiltrating the nasal mucosa of patients with AIA are deficient in EP2 receptor.39
Of 370 single nucleotide polymorphismss from 63 candidate genes, only a particular variant coding for EP2 was significantly associated with AIA.40
The evident discrepancy between the systemic biosynthesis of PGE2, evaluated by its two main metabolites, and accumulating data for PGE2 inhibition as the triggering mechanism of AIA could be explained by a cellular mechanism. Hypersensitivity to aspirin and other drugs of its class is a phenomenon linked to a specific inflammation41 in which there is degranulation of mast cells in target organs such as the bronchial wall, nasal and paranasal mucosa and skin in patients with urticaria. This has been repeatedly documented by increased levels of PGD2 in blood41 42 and urine,43 44 histamine in nasal secretions,45–47 bronchoalveolar lavage fluid48 and tryptase in blood.49 It is plausible that activated mast cells can augment the biosynthesis of both PGE2 and PGD2. However, only PGD2 was reported in relation to the challenge. It is also likely that inflammatory mediators released from the activated mast cells following a positive aspirin challenge could affect PGE2 production through two mechanisms. First, they could upregulate PGE2 biosynthesis in other cells through an intracellular calcium-dependent phospholipase A2. This rise in systemic PGE2 would depend on COX-2 activity and correlate with the number of inflammatory cells. In fact, a selective COX-2 inhibitor reduced urinary levels of both PGE2 metabolites to the same extent in patients with AIA and those with ATA. Second, they could modulate PGE2-related pathways. Indeed, in primary human airway epithelial cells, interleukin-13 depresses PGE2 production but, at the same time, upregulates the activity of enzymes that metabolise PGE2.50 The activity of the enzymes involved in PGE2 degradation could therefore be upregulated by some of the numerous proinflammatory substances that are released during an aspirin-induced asthma attack. Additional evidence is based on upregulation of the cysLT pathway in AIA. Together with eosinophils, mast cells are the main systemic source of cysLTs. The positive correlation between the post-challenge increase of LTE4 excretion and the blood eosinophil count reflected mast cell- eosinophil functional unit, a hallmark of inflammation associated with cysLT overproduction.51
In this study the urinary LTE4 levels at baseline and following aspirin challenge were in agreement with those shown previously.16 17 However, it was surprising to find a lack of correlation between the urinary PGE2 metabolites and LTE4 levels in patients with AIA. Moreover, the dose of aspirin had no effect on the magnitude of the response of either of the PGE2 metabolites and its duration in these patients.
In the ATA group, placebo, aspirin and celecoxib tended to reduce urinary excretion of LTE4. A similar trend was observed following placebo and celecoxib in patients with AIA. These results suggest that, like placebo, aspirin and celecoxib in patients with ATA and celecoxib in patients with AIA do not affect cys-LT metabolism. The tendency of LTE4 to decrease over time following placebo, aspirin and celecoxib administration in patients with ATA and after placebo and celecoxib in those with AIA may reflect the diurnal variation in cys-LT production in the body. However, this observation needs to be confirmed by further studies.
In one of 17 patients with AIA who developed shock following aspirin challenge, celecoxib triggered signs of bronchial obstruction and a rise in urinary LTE4 levels while the remaining patients tolerated celecoxib very well. Special care is therefore needed if celecoxib is to be given to patients with severe aspirin intolerance. Following celecoxib challenge, urinary LTE4 levels decreased in patients with AIA and remained stable throughout the observation period. This result differs from earlier observations.52 Contrary to previous reports,52 there were no changes in urinary LTE4 levels after celecoxib administration in patients with ATA.
In conclusion, this study describes the systemic production of PGE2 in asthma and points to differences between patients with AIA and ATA. The global production of PGE2 remains unaffected by aspirin in patients with AIA compared with patients with ATA. The lack of depression of systemic PGE2 biosynthesis following aspirin challenge in patients with AIA may be a consequence of mast cell activation with a secondary inflammatory response to the released mediators. However, this may not apply to local conditions in the bronchi where aspirin can suppress PGE2 biosynthesis in patients with AIA.
Acknowledgments
The authors thank Professor John McGiff for his valuable comments.
REFERENCES
Supplementary materials
web only appendix 63/1/27
Files in this Data Supplement:
Footnotes
Funding: This work was supported by grant P01/2006/31 from the Polish Ministry of Science.
Competing interests: None.