Article Text

Abstract
Background Rhinovirus (RV) infections are the major precipitant of asthma exacerbations. While neutrophilic lung inflammation occurs during such infections, its role remains unclear. Neutrophilic inflammation is associated with increased asthma severity and steroid refractory disease. Neutrophils are vital for controlling infections but also have immunomodulatory functions. Previously, we found that neutrophils respond to viral mimetics but not replication competent RV. We aimed to investigate if neutrophils are activated and/or modulate immune responses of monocytes during RV16 infection.
Methods Primary human monocytes and autologous neutrophils were cocultured with or without RV16, in direct contact or separated by transwells. RV16-stimulated monocytes were also exposed to lysed neutrophils, neutrophil membrane components or soluble neutrophil intracellular components. Interleukin 6 (IL-6) and C-X-C motif (CXC)L8 mRNA and proteins were measured by quantitative PCR and ELISA at 24 hours.
Results RV16 induced IL-6 and CXCL8 in monocytes, but not neutrophils. RV16-induced IL-6 and CXCL8 from monocytes was reduced in the presence of live neutrophils. Transwell separation abolished the inhibitory effects. Lysed neutrophils inhibited RV16-induced IL-6 and CXCL8 from monocytes. Neutrophil intracellular components alone effectively inhibited RV16-induced monocyte-derived IL-6 and CXCL8. Neutrophil intracellular components reduced RV16-induced IL-6 and CXCL8 mRNA in monocytes.
Conclusions Cell contact between monocytes and neutrophils is required, and preformed neutrophil mediator(s) are likely to be involved in the suppression of cytokine mRNA and protein production. This study demonstrates a novel regulatory function of neutrophils on RV-activated monocytes in vitro, challenging the paradigm that neutrophils are predominantly proinflammatory.
- Asthma
- Respiratory Infection
- Viral infection
- COPD Exacerbations
- Innate Immunity
- Neutrophil Biology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
If neutrophils are meant to control infections but do not respond to rhinovirus, then what is their role during such infections?
What is the bottom line?
Neutrophils are non-responsive to rhinovirus but modulate immune responses of proinflammatory monocytes.
Why read on?
In this study, we describe a novel anti-inflammatory role of neutrophils on rhinovirus-stimulated monocytes. This changes how we view neutrophils during infections and provides valuable insights to their role during viral-induced asthma exacerbations.
Introduction
Neutrophils are the most abundant immune cell in the circulation and play a vital role in innate immune responses. These cells are the first line of defence with the ability to quickly migrate from the circulation to the site of infection. Neutrophils are short-lived terminally differentiated cells that mediate acute anti-microbial functions through the action of cytotoxic granule proteins and reactive oxygen species (ROS). They are phagocytic and take up pathogens into the phagosome where granules filled with toxic compounds fuse with the phagosome to eliminate the microbes.1 Degranulation is the other major effector function where neutrophils exocytose granules from the cytoplasm. Important neutrophil products include neutrophil elastase (NE) which cleaves microbial virulence factors,1 matrix metalloproteinase (MMP)-9 that digests proteases that degrade NE2 and ROS which damage DNA and essential proteins.3 Improved neutrophil responses are associated with enhanced viral clearance4 and less severe disease.5
It is becoming evident that neutrophils are more complex than initially thought and have other roles including in the resolution of inflammation6 and regulation of the subsequent adaptive immune response.7 Recently, the interaction between neutrophils and the adaptive immune system has started to be unravelled. For example, interaction of the programmed death receptor 1 (PD-1) and its ligand, PD-L1, along with the subsequent release of ROS from neutrophils inhibited T-cell-derived interferon-γ (IFN-γ).8 Conversely, activated neutrophils can stimulate dendritic cell maturation so that in turn they stimulate T-cell proliferation.9 Neutrophils are also sources of anti-inflammatory mediators such as transforming growth factor (TGF)-β1 and prostaglandins.10
All common respiratory diseases are characterised by periods of stable disease, interspersed by acute episodes of worsening symptoms known as exacerbations.11 Respiratory viral infections are a major precipitant of exacerbations, with detection rates in exacerbating individuals of up to 78% in asthma,12 64% in COPD13 and 46% in cystic fibrosis.14 In particular, rhinoviruses (RV) are the most common virus type detected during exacerbations.15 The reasons why viral infections promote exacerbations remain unclear. In our previous studies, we showed that neutrophils had functional receptors that detect and respond to viral components. However, when challenged with replication competent RV neutrophils did not respond with increased CXCL8, NE or MMP-9.16 In contrast, structural airway cells17 ,18 and monocytes do respond to RV.19
In this study, we characterised the interaction between monocytes, neutrophils and RV. We observed that neutrophils have an immunomodulatory effect on monocytes and inhibit RV-induced cytokine release. We describe a novel anti-inflammatory role for neutrophils when they interact with RV-stimulated monocytes.
Materials and methods
Volunteer recruitment
The project was approved by the Human Research Ethics Committee, The University of Sydney prior to commencement (protocol number: 14813). Volunteers were recruited by approved flyers placed around The University of Sydney campuses. Participants were required to be over 18 years of age and be fluent in English. Exclusion criteria included if they were pregnant, known to faint during venipuncture procedures or had a bloodborne infection or condition. All patients provided written informed consent, and basic demographic information was collected.
Monocyte and neutrophil isolation
Peripheral blood was collected from volunteers, and neutrophils and monocytes were isolated using a modified standard protocol.16 Briefly, whole blood containing acid citrate dextrose as the anti-coagulant was mixed with 10% dextran (MP Biomedicals, Santa Ana, California, USA) to allow for red blood cell sedimentation. The upper layer was removed and overlayed on Ficoll Paque-PLUS (GE Healthcare, Little Chalfont, UK) and centrifuged at 490×g for 10 min. The peripheral blood mononuclear cells at the interface of plasma and Ficoll were isolated, and monocytes were positively selected using magnetic beads against CD14. From the granulocyte pellet, neutrophils were positively selected using magnetic beads against CD16. Further details are provided in the online supplement. Typical purity of both cell types were 99% or greater.
Supplemental material
RV16
RV16 was generously donated by Professor Sebastian Johnston, Imperial College, London. RV16 was grown in HeLa cells by standard procedures.20 RV16 was purified from viral stocks generated from HeLa cells by polyethylene glycol precipitation and filtration through a 100 000 molecular weight filter.21 Infectivity titres were determined by a titration assay.20 Purified RV16 was used in all experiments.
Neutrophil and monocyte coculture
Neutrophils and monocytes were resuspended in 1% fetal bovine serum (Glendarach Biologicals, Melbourne, Australia), 1% 1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Gibco) and 1% penicillin/streptomycin roswell park memorial institute (RPMI) 1640 (Gibco). 0.5×106 neutrophils and 0.5×106 of autologous monocytes were cultured separately and together at a 1:1 ratio in 24-well plates. They were cultured in media alone (control) and in the presence of RV16 at a multiplicity of infection (MOI) of 1 infectious particle per cell as previously published.22 Cells were incubated at 37°C with 5% CO2 for 24 hours and cell-free supernatant was collected and stored at −80°C for analysis.
Neutrophil and monocyte coculture with transwell separation
Neutrophils were placed into transwell inserts while autologous monocytes were placed into the well below. The coculture system was modified with 0.4 or 3 μm pore size polyethylene terephthalate (PET) transwell inserts (Corning, Corning, New York, USA). RV16 (MOI1) was placed into the lower chamber at time of cell seeding and cultured at 37°C with 5% CO2 for 24 hours and pooled (upper and lower chamber) cell-free supernatant was collected and stored at −80°C for analysis.
To confirm that neutrophils were able to migrate through the 3 μm pore size PET transwell inserts, neutrophils were placed into the upper chamber. Previously collected cell-free supernatants from RV-stimulated monocytes were placed in the lower chamber. Neutrophils were cultured for 1 hour at 37°C with 5% CO2. Manual cell counts were performed on neutrophils that had migrated into the lower chamber.
Effects of lysed neutrophils and neutrophil components on monocytes
The same direct coculture system was set up; however, live neutrophils were replaced with lysed cells. Neutrophils at the same concentration were frozen at −80°C for 15 min and then thawed at rapidly at 37°C before being resuspended and placed with autologous monocytes with the same protocol in the presence and absence of RV16 (MOI1).
Alternatively, the thawed solution was spun in an Eppendorf centrifuge at 16.2×g for 10 min to pellet the membranes. The supernatant was collected, and the intracellular components of 0.5×106 lysed neutrophils were applied to autologous monocytes with or without RV16 (MOI1). The pellet was washed three times with media and centrifuged for 5 min after each wash. The pellet was resuspended, and the membrane fragments were added to separate monocyte cultures with or without RV16.
For heat inactivation, the intracellular components were heated to 95°C for 10 min and then cooled for 5 min at 4°C before applying to monocyte cultures with or without RV16 (MOI1).
All cultures were incubated at 37°C with 5% CO2 for 24 hours, and cell-free supernatant was collected and stored at −80°C for analysis.
Investigation of immune suppressive role of TGF-β1
Neutralising antibodies specific for TGF-β1 (R&D Systems, Minneapolis, Minnesota, USA) were added to either monocytes alone or coculture of monocytes and neutrophils at a final concentration of 10 μg/mL as previously described.23 The cultures were incubated at 37°C with 5% CO2 for 24 hours, and cell-free supernatant was collected and stored at −80°C for analysis. Neutralising antibodies specific for TGF-β1 were confirmed to effectively neutralise recombinant TGF-β1 (see online supplementary figure S1).
Recombinant TGF-β1 (R&D Systems) was added to monocytes alone at 4 and 20 μg/mL and incubated at 37°C with 5% CO2 for 24 hours, and cell-free supernatant was collected and stored at −80°C for analysis.
Effect of indomethacin treatment on monocytes
Neutrophils, monocytes and the combination of neutrophils and monocytes were pretreated with 10−6 M indomethacin (Sigma Aldrich) as previously published,24 dimethyl sulfoxide (DMSO) (Sigma Aldrich) as a vehicle control or no treatment (control) for 30 min at room temperature. Cells were then incubated at 37°C with 5% CO2 for 24 hours in the presence or absence of RV16 (MOI1). Cell-free supernatant was collected and stored at −80°C for analysis. 10−6 M indomethacin was confirmed to be an effective concentration to prevent PGE2 synthesis (see online supplementary figure S2).
Effect of H2O2 on monocytes
Monocytes were treated with increasing concentrations of H2O2 (0.0125 nM, 0.125 nM, 0.25 nM and 100 µM) (Sigma Aldrich). Monocytes with no treatment and monocytes cocultured with neutrophils were used as controls. Cells were treated for 24 hours at 37°C with 5% CO2. Cell-free supernatant was collected and stored at −80°C for analysis.
Effect of neutralising PD-1 on monocytes
Neutrophils, monocytes and the combination of neutrophils and monocytes were pretreated with 10 μg/mL of neutralising PD-1 antibodies as previously described25 or isotype control (BioLegend, San Diego, California, USA) at 37°C for 1 hour. Cells were then either stimulated with RV16 (MOI1) or left unstimulated and incubated at 37°C with 5% CO2 for 24 hours. Cell-free supernatant was collected and stored at −80°C for analysis.
Quantitative PCR
Interleukin 6 (IL-6), CXCL8, IFN-α, IFN-β, IL-28, IL-29A, TNF-α and vascular endothelial growth factor (VEGF) mRNA expression in monocytes were detected using quantitative PCR (qPCR). Total cellular RNA was isolated using the NucleoSpin RNA kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instructions. RNA was converted to cDNA (Invitrogen, Carlsbad, California, USA), and qPCR was carried out on cDNA using the StepOne Real-Time PCR system (Applied Biosystems, Carlsbad, California, USA) using Taqman Gene Expression Assays (Applied Biosystems). Further details can be found in the online supplement.
ELISAs
IL-6, CXCL8 and TGF-β1 production was measured using a sandwich ELISA in duplicate. Specific ELISA kits from R&D Systems (Minneapolis, Minnesota, USA) were used according to the manufacturer’s instructions. Detection limits were 15.6 pg/mL for IL-6 and 31.2 pg/mL for CXCL8 and TGF-β1.
Statistical analysis
Data are presented as the mean±SEM. Data were assumed to be non-parametric due to small sample sizes. A non-parametric Wilcoxon paired t-test or a non-parametric paired one-way analysis of variance with Dunn's multiple comparison test was performed. Significant changes were identified where (p<0.05).
Results
Monocytes but not neutrophils are stimulated to produce cytokines by RV16
We first assessed the production of cytokines by neutrophils and monocytes exposed to RV16 at a range of MOIs (0.5–5) and found that an MOI1 was the lowest concentration to illicit a response (see online supplementary figure S3). Monocytes had a robust response to RV16 exposure with the induction of both IL-6 and CXCL8 release (figure 1A, B). In contrast, neutrophil IL-6 release was below the detection limit of the assay both basally and with RV16 stimulation. Interestingly, neutrophil CXCL8 basal release was low (244 pg/mL) and was not induced in the presence of RV16 (figure 1C).

RV16 induced cytokine release from human peripheral blood monocytes but not from peripheral blood neutrophils. (A) IL-6 and (B) CXCL8 release from human peripheral blood monocytes, stimulated with RV16 (multiplicity of infection of 1 (MOI1)) for 24 hours (n=14). (C) CXCL8 release from human peripheral blood neutrophils, stimulated with RV16 (MOI1) for 24 hours (n=10). IL-6 release from neutrophils was below the limit of detection and is not shown. Data are presented as mean±SEM. ***p<0.001. IL, interleukin; RV, rhinovirus.
Coculture of monocytes with neutrophils and RV16 exposure results in cell contact-mediated inhibition of cytokine release
As neutrophils did not respond to RV16, we investigated whether monocytes activated with the virus interacted with neutrophils in coculture. Remarkably, monocyte-derived RV16-induced IL-6 and CXCL8 was reduced by ∼50%–70% in the presence of neutrophils (figure 2A, B).
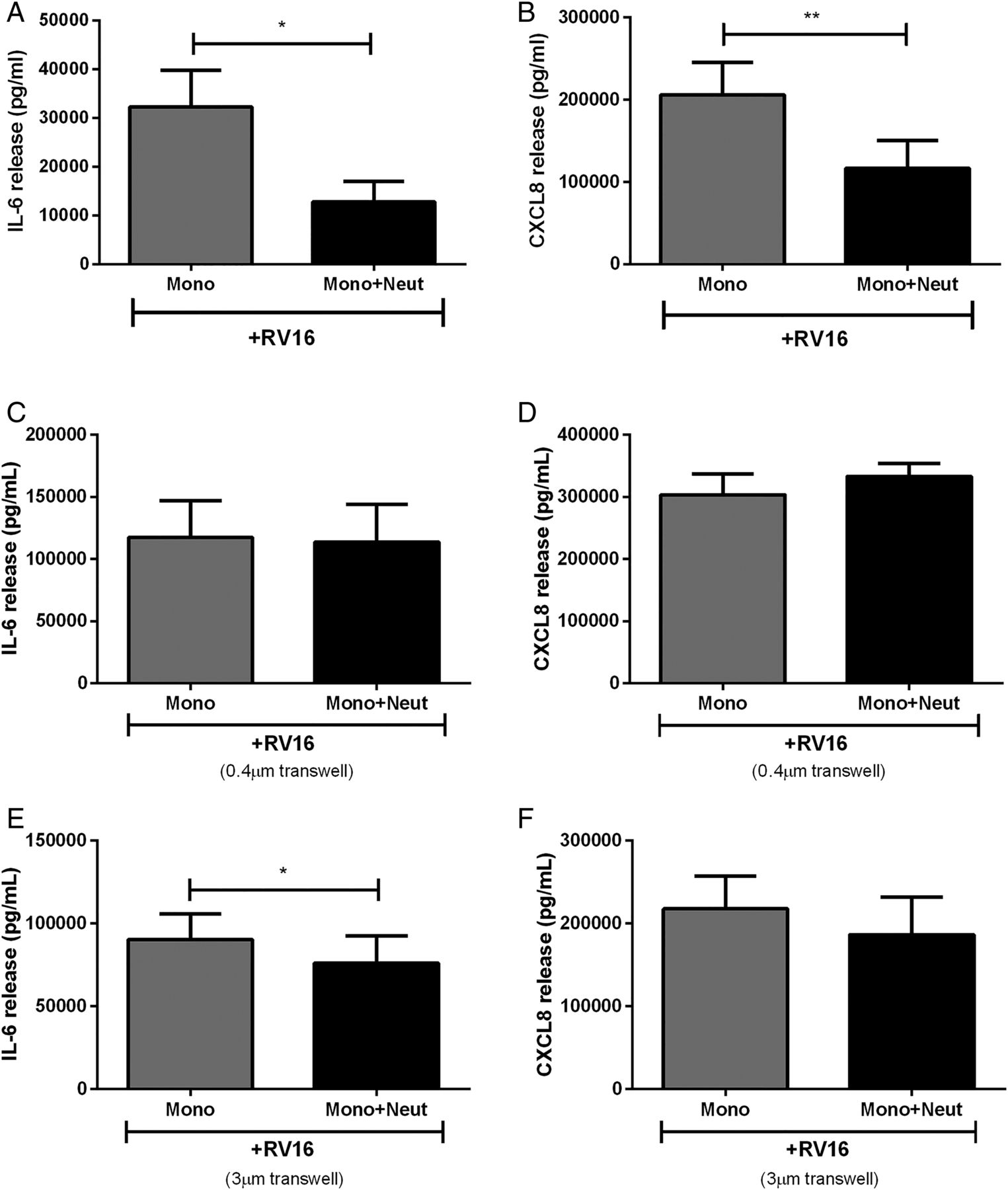
Cytokine release from human peripheral blood monocytes in coculture with autologous neutrophils is altered by cell contact. (A) IL-6 and (B) CXCL8 release from monocytes cocultured with neutrophils, or separated by cell impervious (C and D) or cell porous (E and F) transwells and stimulated with RV16 (multiplicity of infection of 1) (n=5–10). Data are presented as mean±SEM. *p<0.05, **p<0.01 between indicated groups. IL, interleukin; RV, rhinovirus.
The interaction between cocultured monocytes and neutrophils may involve contact between the two cell types and/or the release of soluble factors. To assess these possibilities, we modified the coculture model. Monocytes were separated from neutrophils with 0.4 μm transwells where cells were unable to pass through the membranes but soluble mediators were freely able to diffuse. Prevention of cell-to-cell contact resulted in the loss of neutrophil inhibitory effects on monocyte cytokine release (figure 2C, D).
We also increased the transwell pore size to 3 μm to allow neutrophils in the upper chamber to migrate towards monocytes in the lower chamber in an attempt to restore the inhibitory response. We initially confirmed that neutrophils migrated through the transwell towards the supernatant from RV16-stimulated monocytes over the course of 1 hour (control: 50×104 cells/mL, RV-stimulated monocyte supernatant: 127×104 cells/mL, data not shown). However, over the course of 24 hours the inhibitory effects on CXCL8 release were unable to be rescued when neutrophils were allowed to migrate towards monocytes but were slightly restored with IL-6 release (figure 2E, F).
PD-1 does not appear to mediate inhibition of monocyte-derived cytokine release
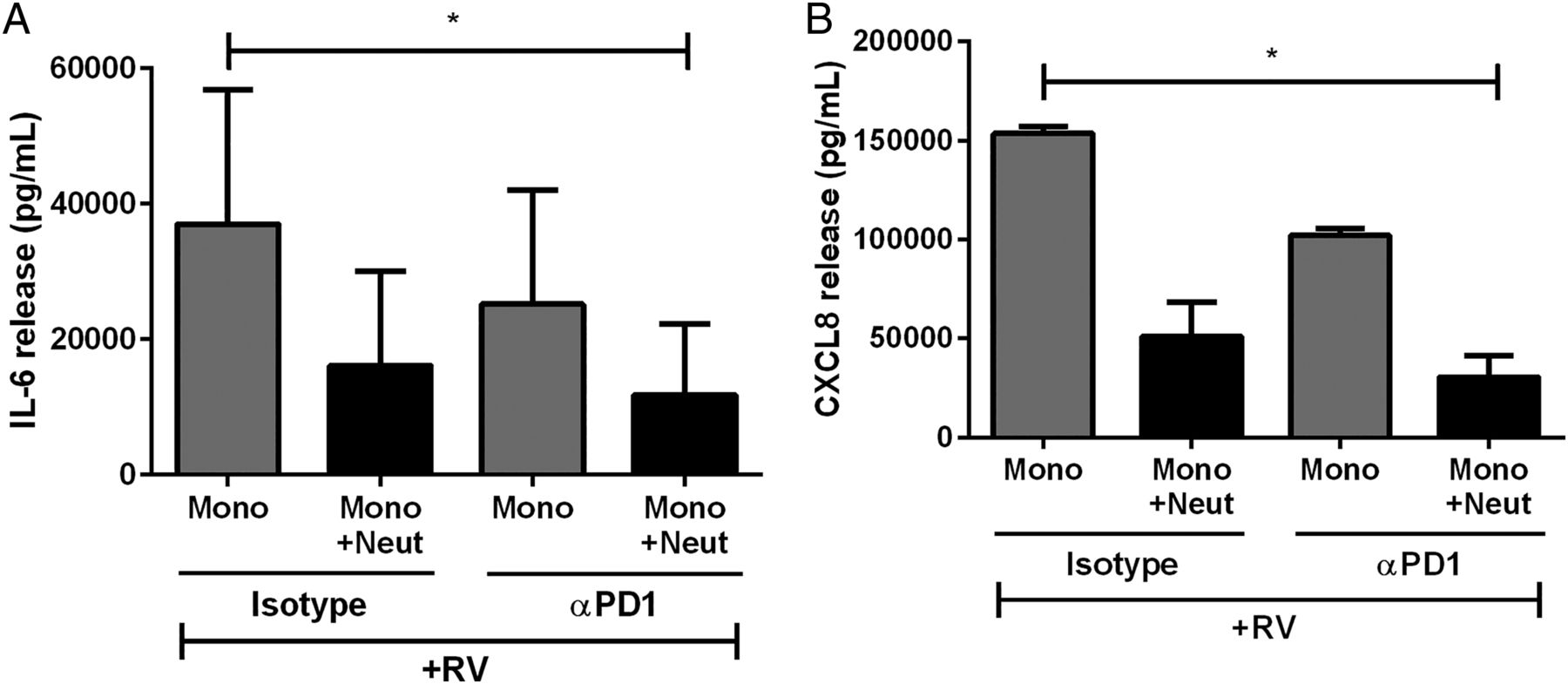
We then investigated the mechanisms of cell contact-mediated inhibition of cytokine release. Since de Kleijn et al8 found that PD-1/PD-L1 ligation had anti-inflammatory effects in T cells, we investigated if PD-1/PD-L1 was responsible for neutrophil inhibition of virus-activated monocytes. There was no difference in cytokine release in the presence or absence of the anti-PD-1 antibody (figure 3A, B). Thus, the ligation of these cell surface proteins does not appear to be essential for neutrophil inhibition of monocyte-derived cytokines.

The effect of programmed death receptor 1 (PD-1) inhibition on cytokine release from human peripheral blood monocytes. (A) IL-6 and (B) CXCL8 release from monocytes cultured alone or with neutrophils with isotype control antibodies or with αPD-1 neutralising antibodies and stimulated with RV16 (multiplicity of infection of 1) (n=3). Data are presented as mean±SEM. *p<0.05. IL, interleukin; RV, rhinovirus.
A constitutively produced neutrophil factor inhibits RV16-induced monocyte-derived cytokine release by reducing monocyte mRNA levels
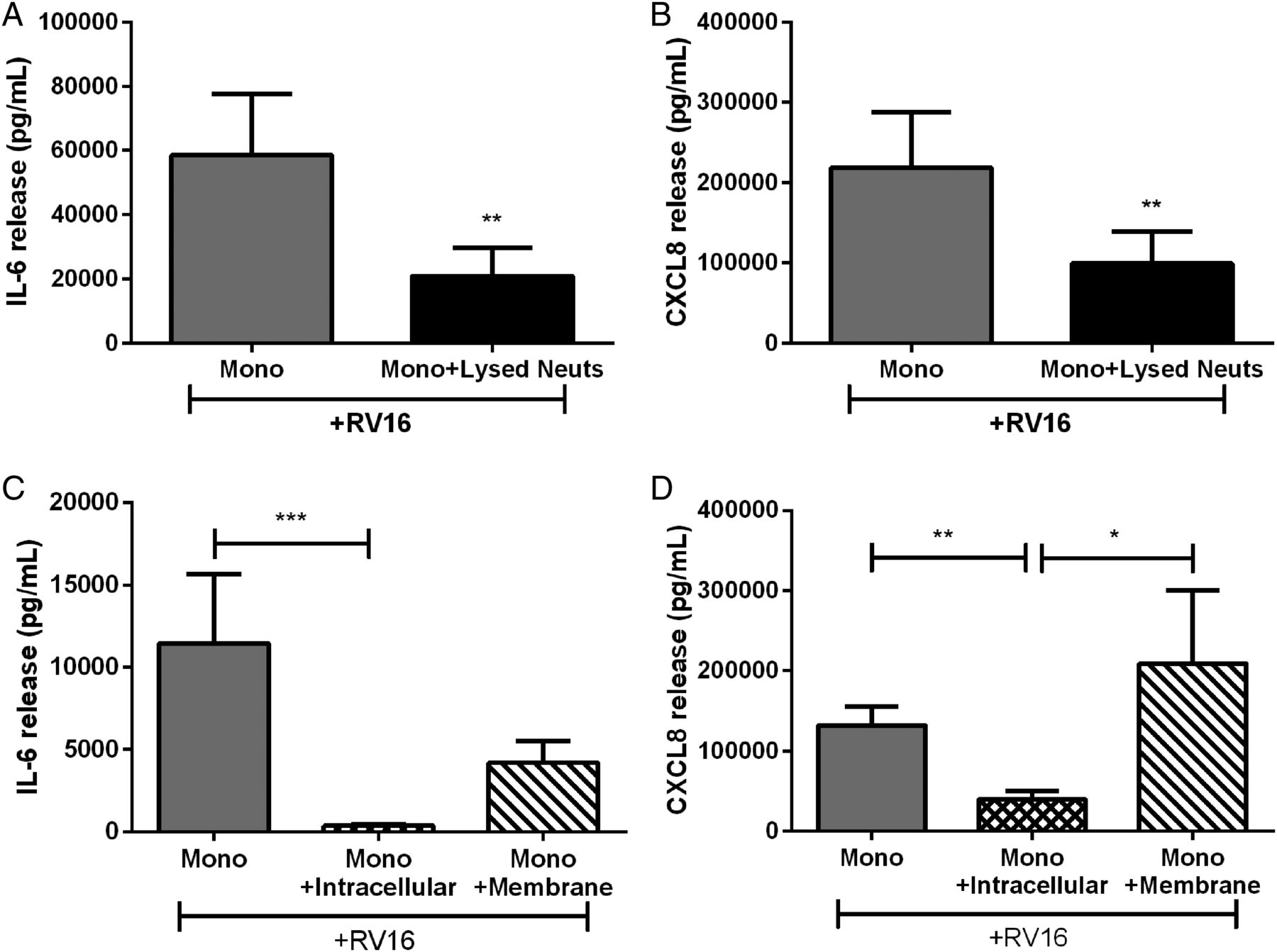
As inhibition of RV16-induced monocyte cytokine release was through cell contact, we investigated if neutrophils constitutively modulated the immune response or if de novo protein was required. We treated autologous monocytes with lysed neutrophils (the same concentration as previous experiments). Lysed neutrophils inhibited RV-induced IL-6 and CXCL8 release to the same degree as in the whole cell coculture model (figure 4A, B).

Cytokine release from human peripheral blood monocytes cultured with autologous lysed neutrophils or components of neutrophils. (A) IL-6 and (B) CXCL8 release from monocytes cocultured with lysed neutrophils or intracellular and membrane components of neutrophils (C and D) and stimulated with RV16 (multiplicity of infection of 1). Data are presented as mean±SEM. (n=8–10). *p<0.05, **p<0.01, ***p<0.001 between indicated groups. IL, interleukin; RV, rhinovirus.
We then expanded this analysis by separating the lysed neutrophil solution into the membranous and soluble intracellular components to elucidate if inhibition was mediated by membrane bound or intracellular factors. The intracellular component effectively inhibited RV16-induced monocyte IL-6 and CXCL8. While the membrane component caused a 60%–70% reduction in IL-6, it was not statistically significant (figure 4C). The membrane component did not inhibit RV-induced CXCL8 release from monocytes (figure 4D). As the neutrophil intracellular component consistently inhibited RV-induced IL-6 and CXCL8 from monocytes, subsequent experiments focused on the inhibitory effects of the neutrophil intracellular component.
We then examined whether the effects occur at the gene transcription level. We assessed cytokine mRNA levels in monocytes treated with neutrophil intracellular components and stimulated with RV16. We found that IL-6, CXCL8 and TNF-α mRNA were downregulated following exposure to neutrophil intracellular components while IFNα and IFNβ were not altered (figure 5A–E). IL-28, IL-29A and VEGF mRNA were not detected.
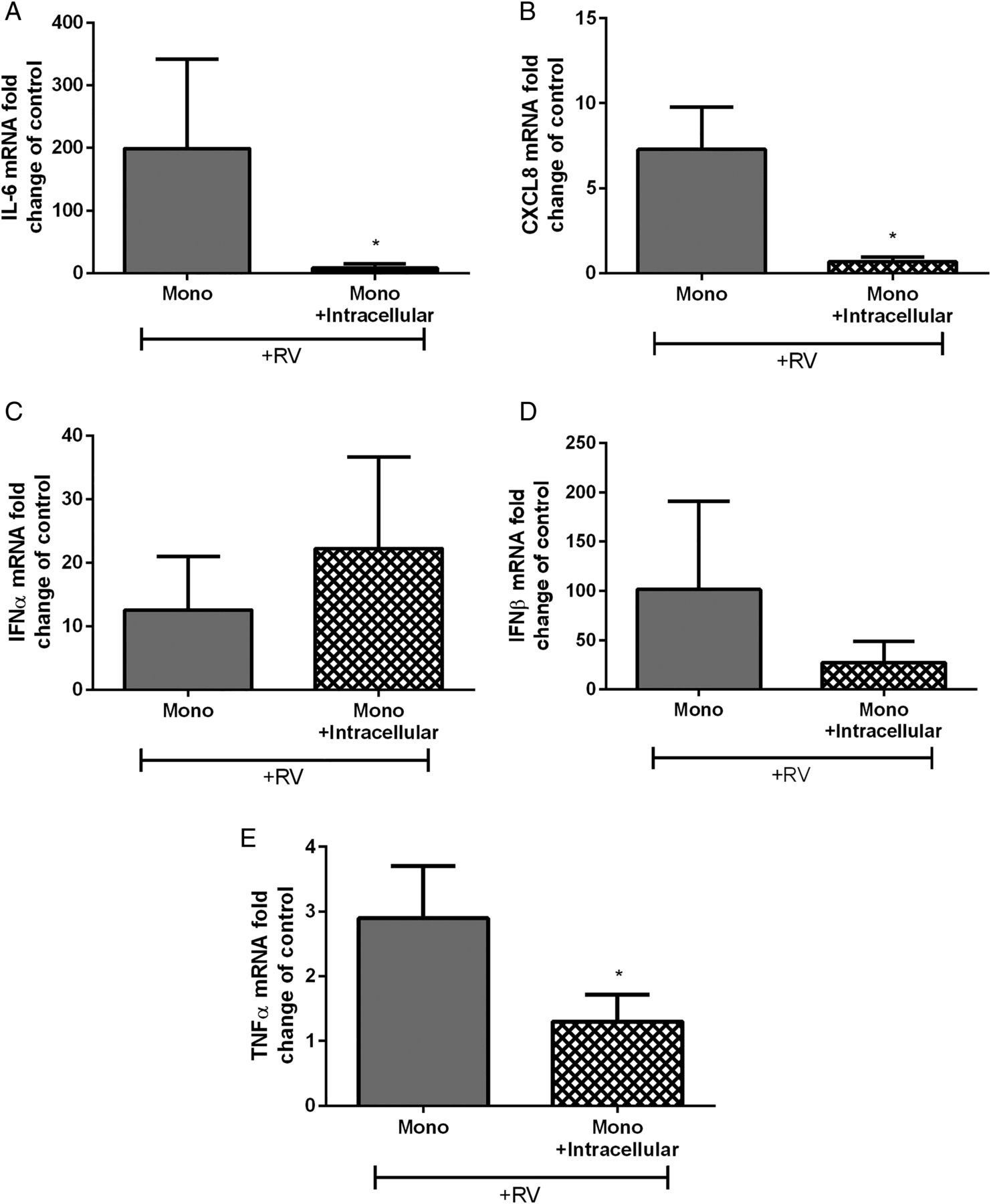
Cytokine mRNA expression in human peripheral blood monocytes cultured with isolated autologous neutrophil intracellular components. (A) IL-6, (B) CXCL8, (C) IFN-α, (D) IFN-β and (E) TNF-α monocyte mRNA after RV16 (multiplicity of infection of 1) stimulation represented as fold change of unstimulated control (n=5–7). Data are presented as mean±SEM. *p<0.05. IL, interleukin; RV, rhinovirus.
TGF-β1 is unlikely responsible for neutrophil inhibitory effects
TGF-β1 has known anti-inflammatory effects and was present in supernatants from cocultured monocytes and neutrophils (figure 6A, B). However, RV16 did not induce TGF-β1 from neutrophils (figure 6C) or monocytes (figure 6D). To investigate the role of TGF-β1 further, we examined the effect of neutralising antibodies in the coculture model. Depletion of TGF-β1 did not affect the inhibition of IL-6 or CXCL8 production from RV-activated monocytes by neutrophils (figure 6E, F). Furthermore, addition of recombinant TGF-β1 (4 and 20 ng/mL) reduced IL-6 release by 50% (not statistically significant) compared with monocytes alone but was not as effective as neutrophils cultured with monocytes (figure 6G). However, it did not prevent the inhibition of CXCL8 release (figure 6H).

The effect of transforming growth factor (TGF)-β1 on cytokine release from human peripheral blood monocyte and neutrophil cocultures. (A) TGF-β1 release from monocytes alone and cocultured with neutrophils in the absence of RV16 and (B) in the presence of RV16 (multiplicity of infection of 1 (MOI1)) (n=6–7). (C) TGF-β1 release from neutrophils and (D) monocytes stimulated with RV16 (MOI1) (n=5–6). (E) IL-6 and (F) CXCL8 release from monocytes and neutrophil cocultures in the presence of anti-TGF-β1 neutralising antibodies and stimulated with RV16 (MOI1) (n=4). (G) IL-6 and (H) CXCL8 release from monocytes stimulated with recombinant human TGF-β1 (n=6). Data are presented as mean±SEM. *p<0.05, **p<0.01. IL, interleukin; RV, rhinovirus.
Neutrophil soluble factor is heat resistant and is unlikely to be a prostaglandin or H2O2
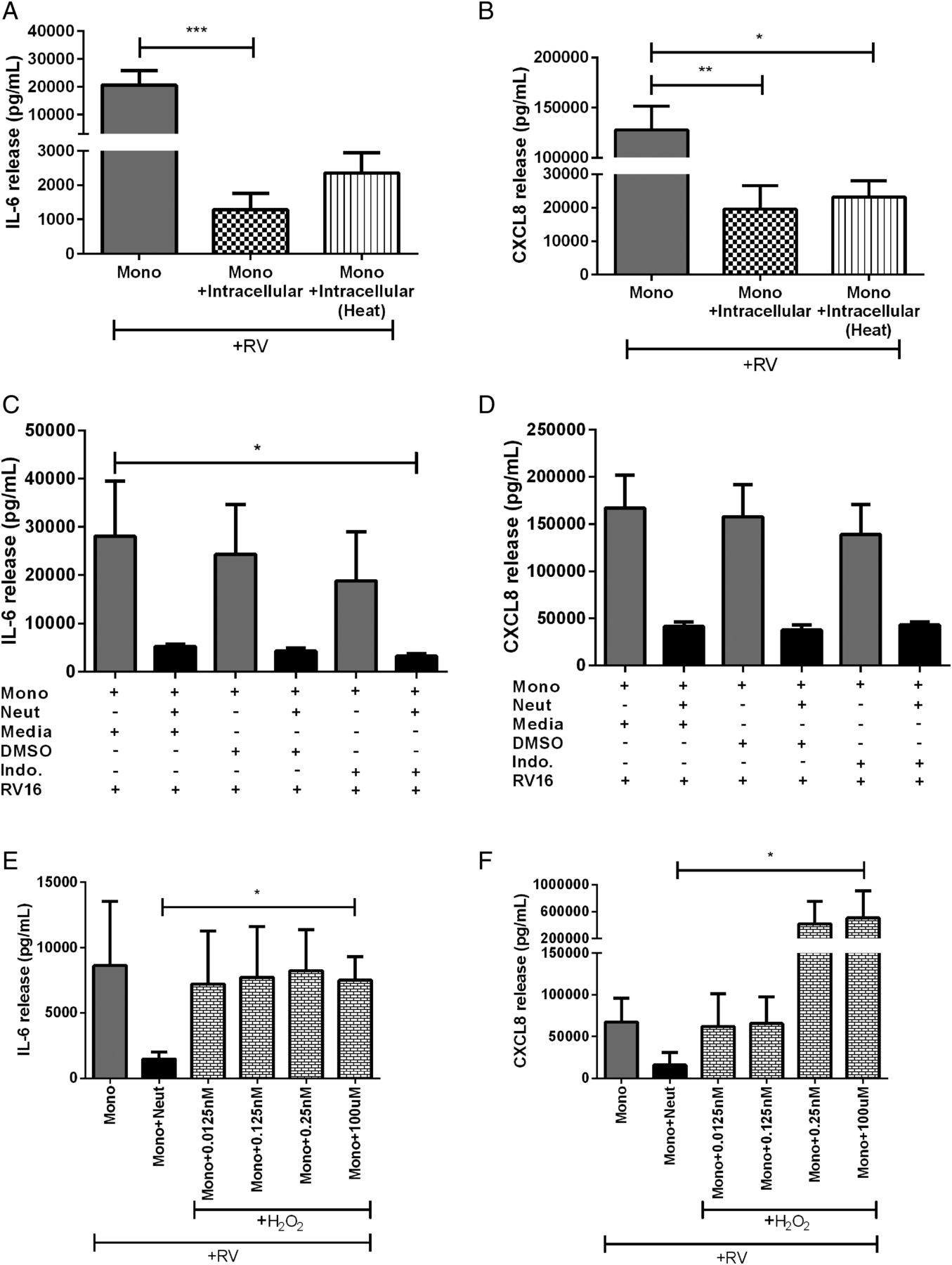
It is unlikely that TGF-β1 is the soluble neutrophil factor responsible for inhibiting RV-activated monocytes as it appeared to trend towards a decrease in IL-6 but not CXCL8. We then further characterised the neutrophil intracellular component in attempts to identify the molecule responsible. As most proteins can be denatured by heat, and hence lose activity, we investigated if the inhibitory factor is heat labile. Heating did not abolish the inhibitory effects of the intracellular neutrophil components on IL-6 or CXCL8 release (figure 7A, B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The effect of neutrophil-related factors on cytokine release from human peripheral blood monocyte and neutrophil cocultures. (A) IL-6 and (B) CXCL8 release from monocytes cultured with autologous heat-inactivated neutrophil intracellular components and in the presence of RV16 (multiplicity of infection of 1 (MOI1)) (n=9). (C) IL-6 and (D) CXCL8 release from monocytes and monocytes cocultured with neutrophils in either media alone, DMSO vehicle control or indomethacin or (E and F) in the presence of H2O2 and stimulated with RV16 (MOI1) (n=3–4). Data are presented as mean±SEM. *p<0.05, **p<0.01, ***p<0.001. IL, interleukin; RV, rhinovirus.
Since, it appears that a protein is not the unknown inhibitory factor isolated from neutrophils, we next examined the potential for it to be a lipid mediator, such as prostaglandins, as they can have anti-inflammatory functions.26 We treated RV-activated monocytes and neutrophils in coculture with the cyclooxygenase (COX) inhibitor, indomethacin (figure 7C, D). We found no difference with treatment, indicating that prostaglandins or other lipid mediators are unlikely to be involved.
Previously, Pillay et al27 have described a role for neutrophil-derived H2O2 in inhibiting T-cell proliferation. We next explored the possibility for neutrophil-derived H2O2 to be the inhibitory factor in our model by adding it into monocyte cultures. As expected, the coculture of monocytes with neutrophils had around a 75% decrease in IL-6 and CXCL8 release compared with monocytes alone. The addition of H2O2 to monocytes alone resulted in no change in IL-6 release but a dose-dependent increase in CXCL8 (figure 7E, F). However, we were unable to detect H2O2 in the supernatants from neutrophils and monocytes stimulated with RV16 nor the coculture of the two cell types using a fluorescence assay kit (Cayman Chemicals, Ann Arbor, Michigan, USA).
Discussion
To our knowledge, this is the first study to observe an immunomodulatory function of neutrophils on monocyte function in the context of an RV infection. We found that neutrophils are unable to respond directly to RV16 with cytokine release but monocytes are stimulated to release large amounts of IL-6 and CXCL8 by the virus. Neutrophils appear to be non-responsive to RV16 but actually reduce cytokine release from activated monocytes in a cell contact-dependent manner that is unlikely to be PD-1/PD-L1 dependent. A soluble factor that is constitutively produced in neutrophils is responsible; this soluble factor(s) reduces cytokine mRNA production and in turn protein production in monocytes. We have deduced that this unknown neutrophil factor resides internally, is resistant to heat and is unlikely to be TGF-β1, a lipid mediator such as prostaglandins or H2O2.
Neutrophils are thought to be a homogenous population of effector, proinflammatory cells. They are known to migrate swiftly to sites of infection to contain the spread of the pathogen. We know that neutrophils migrate to the airways during a respiratory viral infection28 and that improved neutrophil responses help control infections4 and limit the development of severe disease.5 Neutrophilia occurs in stable asthmatic patients 29 and in patients with COPD,30 and in experimental asthma and COPD neutrophilia is associated with more severe, steroid-insensitive disease.31–33 However, the role of the neutrophil in clearing RV is not clear despite their influx and this raises the question of their purpose during such infections. Further clinical and experimental studies using appropriate models are required to fully elucidate their roles.34 ,35
Our previous work has shown that despite neutrophils expressing functional innate immune receptors for detecting RV, they are non-responsive to live RV16.16 Neutrophil activation appears to be due to inflammatory mediators generated from other cells.36 We know that the primary site of RV infection is the epithelium37 ,38 and these cells could be the source of mediators that activate neutrophils. However, our preliminary work suggested otherwise. We applied RV16-infected BEAS-2B supernatants onto neutrophils. We also cocultured neutrophils with RV16-infected primary bronchial epithelial cells. In both cases, we found that neutrophils were not activated. This led us to suspect that perhaps another immune cell capable of detecting the presence of RV16 could in turn activate neutrophils.
Monocytes are bone marrow-derived cells that are predominately found in the circulation. They can differentiate into dendritic cells and a variety of tissue specific macrophages, and mediate host defence. Like most leucocytes, monocytes migrate to the airways in response to signals from the epithelium,39 through adhesion to the endothelium, tethering and extravasation following a chemotactic gradient, usually CCL2 monocyte chemoattractive protein 1 (MCP1).40
Interestingly, our preliminary work showed that monocytes produce approximately 10 times more CXCL8 in response to RV16 compared with lipopolysaccharide (LPS)-stimulated neutrophils. CXCL8 is a potent neutrophil chemoattractant, and monocytes are a significant source of this chemokine. The new immunomodulatory functions of neutrophils that we have identified in this study may be a feedback mechanism to control these monocyte-driven inflammatory responses, which when excessive may be tissue damaging. While our study focused on the mechanism of inhibition, future studies looking at whether there is dysfunction in this mechanism in asthmatic, COPD or cystic fibrosis (CF) neutrophils will be of substantial interest and may potentially be manipulated for therapeutic benefit. In addition, as monocytes have the potential to differentiate into macrophages and dendritic cells, it would also be important to investigate if neutrophils also have immunomodulatory effects on these cells.
TGF-β1 was detected in our coculture supernatants and has known anti-inflammatory effects,41 however, was not induced by RV16. Nevertheless, antibody neutralisation in the coculture model along with the addition of TGF-β1 to monocytes alone at concentrations greater than we measured in our coculture supernatants excluded this cytokine from being the soluble factor we were searching for.
We also assessed the potential role of PD-1/PD-L1 and H2O2 to be involved in this mechanism as previous reports have implicated these factors in suppressing T-cell activity.8 ,27 Our data are limited in that we were unable to provide positive controls in our PD-1/PD-L1 experiments. However, our data suggest that it is unlikely that PD-1/PD-L1 is involved but further investigation is required. In addition, we only investigated the effect of H2O2 on monocyte cultures and not on the coculture model. Nevertheless, we conclude that exogenous H2O2 appears to have no effect on RV-induced cytokine release from monocytes and therefore is also unlikely to be the mediator in question. There remains the potential that a different receptor(s) is/are engaged and different neutrophil mediator(s) is/are involved which requires further investigation.
It is becoming clear that the pool of neutrophils is a heterogeneous population. Pillay et al27 described three distinct populations of neutrophils in the circulation after intravenous LPS administration in humans. Neutrophils with single banded nuclei which were likely immature cells released from the bone marrow in response to the stimulus, mature neutrophils with typical segmented nuclei and a third group with hypersegmented nuclei which they found to hold immunomodulatory functions. Interestingly, these suppressor neutrophils and mature neutrophils were CD16high. Our protocol used CD16 magnetic beads to purify the neutrophil samples by positive selection. We may have preferentially selected out CD16high suppressor neutrophils unintentionally during purification from the granulocyte fraction; however, a more detailed investigation of different populations of suppressor neutrophils is outside the scope of this study.
It is evident that neutrophil functions have been oversimplified in the past. It is now clear that they bridge the gap between the innate and adaptive immune system by cross-talking with dendritic cells, B cells and T cells and modulating their responses.42 ,43 Most of the functions of suppressor neutrophils have been described using murine models; however, the few studies conducted in humans align closely with our proposed mechanism. In line with other studies, we found that there is a requirement for cell contact, or a formation of an immunological synapse between the two cell types for inhibition to occur.8 ,9 ,27 However, our experiments suggest that PD-1 and PD-L1 were not involved. Similarly, roles for a secreted molecule into the synapse have been identified, arginase-1 in mice44 and H2O242 in human T-cell models; however, our data suggest that H2O2 is unlikely involved in this mechanism we describe.
This study has limitations as it is an ex vivo model which may not accurately reflect physiological conditions. This model uses 1:1 ratios of neutrophils to monocytes which is possible to occur in vivo during inflammation when both cell types are recruited into the airways; however, it is possible that there may be more monocytes to neutrophils or vice versa. There is also evidence that there are subtypes of neutrophils27 and monocytes45 which we did not characterise in this study. Future studies investigating the proportion and roles of various subtypes of neutrophils and monocytes, particularly suppressor subtypes, would be of interest.
In this study, we routinely used the control conditions of RV-stimulated monocytes alone and cocultured with neutrophils. Despite keeping the protocol consistent between experiments, there are fluctuations in the amounts of IL-6 (10 000–100 000 pg/mL) and CXCL8 (100 000–300 000 pg/mL) induced from monocytes cultured alone. This also occurred with the coculture of monocytes with neutrophils. We suspect the variability is most likely due to the heterogeneity of responses observed when using primary human cells.
We investigated if nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) was dysregulated in monocytes treated with neutrophil products, but found no evidence of NF-κB activation following RV exposure of monocytes (data not shown). From our previous work in airway smooth muscle cells, RV induced IL-6 in an NF-κB dependent pathway.17 This was replication independent, and likely triggered by RV engagement with intracellular adhesion molecule (ICAM)-1. However, in monocytes, RV-induced IL-6 and CXCL8 may occur following viral detection, as RV does not replicate in monocytes46 but RV RNA can be isolated in the monocytes following RV exposure. Therefore, viral recognition through toll like receptor (TLR)3, 7 and/or 8 is likely to occur. Activation of these pathways leading to cytokine production is known to be NF-κB independent. Alternatively, RV-induced cytokine release may differ from other cell types, and still be NF-κB independent even if viral replication occurs within these cells. The lack of NF-κB activation may be beneficial in these cases as it may be detrimental to overly activate cytokine responses (in the worst case leading to a cytokine storm) each time a person suffers from a cold.
In this study, we could not identify the specific inhibitory mechanism involved. We speculate that it could potentially be microRNAs or long intervening non-coding RNAs that may be responsible. MicroRNAs have recently been shown to alter macrophage function,47 and a number have been identified to modulate immune responses and some have been associated with respiratory disease.48
Our research challenges the notion that neutrophils are responsible for aberrant inflammation found during RV infections in asthmatics which results in acute symptoms. We propose that potentially the immunomodulatory functions of neutrophils from asthmatics may be dysfunctional or that monocytes contribute more to the inflammatory environment in these patients than neutrophils. While it is speculation, we believe that it is an important area that future research needs to address.
References
Footnotes
Contributors FSMT, PMH, AJA, JKB, KJB and BGO provided conception and design of the study. FSMT carried out recruitment, completed all cell biology, laboratory work and data analysis. All authors contributed to the preparation of the manuscript.
Funding This study was funded by the National Health and Medical Research Council (NHMRC), Australia. PMH was supported by an NHMRC Principal Research Fellowship, JKB and BGO were supported by NHMRC Career Development Fellowships #1032695 and #1026880.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Human Research Ethics Committee, The University of Sydney.
Provenance and peer review Not commissioned; externally peer reviewed.