Article Text

Abstract
Background: The relationship between airway structural changes and inflammation is unclear in early cystic fibrosis (CF) lung disease. A study was undertaken to determine changes in airway remodelling in children with CF compared with appropriate disease and healthy controls.
Methods: Bronchoalveolar lavage and endobronchial biopsy were performed in a cross-sectional study of 43 children with CF (aged 0.3–16.8 years), 7 children with primary ciliary dyskinesia (PCD), 26 with chronic respiratory symptoms (CRS) investigated for recurrent infection and/or cough and 7 control children with no lower airway symptoms. Inflammatory cells, cytokines, proteases and matrix constituents were measured in bronchoalveolar lavage fluid (BALF). Reticular basement membrane (RBM) thickness was measured on biopsy specimens using light microscopy.
Results: Increased concentrations of elastin, glycosaminoglycans and collagen were found in BALF from children with CF compared with the CRS group and controls, each correlating positively with age, neutrophil count and proteases (elastase activity and matrix metalloproteinase-9 (MMP-9) concentration). There were significant negative correlations between certain of these and pulmonary function (forced expiratory volume in 1 s) in the CF group (elastin: r = −0.45, p<0.05; MMP-9:TIMP-1 ratio: r = −0.47, p<0.05). Median RBM thickness was greater in the CF group than in the controls (5.9 μm vs 4.0 μm, p<0.01) and correlated positively with levels of transforming growth factor-β1 (TGF-β1; r = 0.53, p = 0.01), although not with other inflammatory markers or pulmonary function.
Conclusions: This study provides evidence for two forms of airway remodelling in children with CF: (1) matrix breakdown, related to inflammation, proteolysis and impaired pulmonary function, and (2) RBM thickening, related to TGF-β1 concentration but independent of other markers of inflammation.
Statistics from Altmetric.com
Newborn infants with cystic fibrosis (CF) have structurally normal airways but, at the time of death or lung transplantation, there is severe airway destruction and extensive bronchiectasis.1 It has been assumed that these structural airway wall changes have occurred secondary to infection and inflammation. In asthma, airway remodelling has been thought to follow chronic airway inflammation,2 but recent evidence has challenged this assumption, suggesting instead that remodelling may be an independent parallel process.3 Whether remodelling in CF is secondary to infection and inflammation or a separate process is of potential importance; if the former, then treatment of infection and inflammation could preserve airway function. But if remodelling is a separate process, relating to some aspect of cystic fibrosis transmembrane regulator (CFTR) dysfunction, new therapeutic approaches to preserve airway function may be required.
Neutrophilic inflammation within the airway lumen in CF is central to the pathophysiology of the disease and can occur within the first few months of life.4 However, most previous work on airway wall pathology has come from explanted lungs or at necropsy. Little is known about the nature of histological changes in the airway wall in children with relatively mild disease or early stage disease, and how these changes may relate to inflammation within the airway lumen.
Investigation of airway remodelling in CF to date has attempted to establish alterations in airway components, cytokines and proteases that appear important in asthma. Although there may be airway smooth muscle hyperplasia in adult CF biopsies compared with controls,5 it is unclear whether there is thickening of the reticular basement membrane (RBM) and whether any of these changes are seen in the paediatric age group.6 7 However, biopsy immunoreactivity against transforming growth factor-β1 (TGF-β1), a cytokine known to be pro-fibrotic in vitro,8 appeared to be associated with a better clinical picture.7 Increased levels of matrix metalloproteinase-9 (MMP-9) have been found in sputum and bronchoalveolar lavage fluid (BALF) from children with CF.9 10 Furthermore, increased breakdown products of airway matrix components have been found in urine11 and sputum12 of patients with CF, but with no clear relationship with pulmonary function.
We have previously shown that infants with CF have impaired lung function at diagnosis, irrespective of current or previous respiratory problems,13 and that there is no catch-up in lung function in the preschool years, despite intensive treatment in specialist centres.14 We therefore hypothesised that structural airway wall changes are present early in the course of CF and that, as with atopic asthma, they may not be directly related to infection or inflammation. To this end, we compared endobronchial biopsies and BALF from children with CF with those from disease and healthy control groups. We studied matrix degradation and thickening of the RBM and looked for relationships with markers of airway inflammation and infection.
METHODS
Subjects
Subjects were prospectively recruited from the following four groups of children: (1) CF, diagnosed using standard criteria,15 who were undergoing bronchoscopy for a clinical indication or having a general anaesthetic for another clinically indicated procedure; (2) primary ciliary dyskinesia (PCD)16 who were undergoing bronchoscopy during a pulmonary exacerbation; (3) chronic respiratory symptoms (CRS) who were being electively investigated for a history of recurrent infections and/or persistent or recurrent cough; (4) controls with no history of lower airway problems (3 had recurrent croup, 1 had previous self-reported haemoptysis, 1 had stridor, and 2 were undergoing cardiac catheterisation where informed consent was given to perform bronchoscopy for research reasons).
Before bronchoscopy, spirometry was performed according to American Thoracic Society guidelines17 with a Vitalograph 2120 spirometer (Vitalograph, Ennis, Ireland) and reference data ERS/Polgar. The study was approved by the research ethics committees of the Royal Brompton and Chelsea & Westminster Hospitals. Fully informed consent was obtained from parents and age-appropriate assent from the children.
Bronchoscopy
All bronchoscopies were performed under general anaesthesia. Bronchoalveolar lavage was performed using three aliquots of 1 ml/kg of room temperature 0.9% saline instilled separately into the right middle lobe, unless otherwise indicated, and the return mixed. Up to three endobronchial biopsies were taken under direct vision from subcarinae of segmental bronchi in the right lower lobe with FB-56D-1 rat tooth or FB-231D oval fenestrated jaw forceps (Olympus, Tokyo, Japan), depending on the size of the bronchoscope, and placed in 10% formalin in saline.
BALF analysis
BALF was kept on ice and processed within 2 h. Quantitative microbiology was performed on chocolate, Colombia Blood Agar, Saboraud, Mannitol Salt Agar, Maconkey, Pseudomonas isolation and Burkholderia cepacia media. Viruses (respiratory syncytial virus, parainfluenza 1, 2 and 3, influenza A and B, adenovirus and cytomegalovirus) were detected using direct immunofluorescence and/or rapid viral tissue culture. Cell counts were performed using a dual chamber Neubauer haemocytometer (Assistent, Sondheim, Germany). Remaining BALF was centrifuged at 2000 g for 10 min, the supernatant removed and stored in aliquots at −80°C for later analysis. The cell pellet was then washed, 0.1% dithiothreitol added as a mucolytic agent, washed again and then re-suspended for a cytospin preparation. Cell differential was assessed using May-Grunwald-Giemsa staining and counting of at least 300 identifiable cells.
Interleukin (IL)-8, IL-10, IL-13 and total TGF-β1 were measured using commercial ELISA kits (DuoSet, R&D Systems Inc, Minneapolis, Minnesota, USA). IL-10 was measured with and without protease inhibition (1% 0.5M EDTA (Gibco), 1% protease inhibitor P8340 (Sigma-Aldrich, St Louis, Missouri, USA), 1% BSA (Sigma-Aldrich)). IL-13 and TGF-β1 were measured with protease inhibition. Neutrophil elastase (NE) activity was measured using an assay based on NE cleavage of nitroanilide from N-methoxysuccinyl-ala-ala-pro-val-p-nitroanilide (Sigma-Aldrich). MMP-9 and tissue inhibitor of metalloproteinase-1 (TIMP-1) were measured using Biotrak ELISA kits (Amersham Biosciences, Buckinghamshire, UK). Elastin, glycosaminoglycans and collagen were measured using dye binding assay kits (Biocolor, Newtonabbey, Northern Ireland).
Biopsy analysis
Formalin-fixed paraffin-embedded sections (3–5 μm thickness) were stained with haematoxylin and eosin. One section from each patient was selected which showed identifiable epithelium and submucosa with at least 800 μm of basement membrane. RBM thickness was measured on coded sections using light microscopy and computer-aided image analysis (NIH Image 1.55; National Institutes of Health, Bethesda, Maryland, USA) by taking the geometric mean of 40 measurements at 20 μm intervals, as previously described.18 19
Statistical analysis
Non-parametric tests were applied to test for intergroup differences. Associations were examined by Spearman rank correlation. The χ2 test was used to test for differences in the distribution of categorical variables. A p value of <0.05 was deemed statistically significant, except for multiple comparisons (CF vs each of each different groups) when a p value of 0.017 was used. SPSS V. 11.5 (SPSS Inc, Chicago, Illinois, USA) was used for statistical analysis.
RESULTS
Subjects
The demographic characteristics of the 83 children who underwent bronchoscopy are summarised in table 1. In the CF group, bronchoscopy was performed during a pulmonary exacerbation in 31 children (72%, 26 of whom were receiving intravenous antibiotics) and during a period of clinical stability in 12 (28%). The primary reason for bronchoscopy in the CF group is broken down by age in table 2, with the indications detailed separately for those under and over 6 years of age. It is our routine clinical practice to perform bronchoscopy following diagnosis, and this was the indication in 9 of the 11 very young children (<2 years); bronchoscopy was performed during a pulmonary exacerbation in the other 2 children, one of whom was undergoing line insertion under general anaesthesia. Of the 43 children with CF, 40 had genotypes available; 27 were ΔF508 homozygotes (68%), 10 were compound ΔF508 heterozygotes, 2 were G551D/-, and 1 was N1303K/N1303K.
Bronchoscopy
The right middle lobe was used for bronchoalveolar lavage in 71 cases (86%). Endobronchial biopsies were taken during 78 bronchoscopies (94%); between 1 (4 patients) and 3 (70 patients) biopsies were performed. All patients in the control group, including the one with suspected haemoptysis, had a normal lower airway on bronchoscopy and subsequent negative BALF cultures. No patient had any overt complication from endobronchial biopsy.
BALF analysis
Pathogenic bacteria were found in 13 BALF samples from the CF group (30%), including those from 8 of the 26 children (31%) on intravenous antibiotic therapy, 4 (57%) in the PCD group and 12 (46%) in the CRS group. Pseudomonas aeruginosa was detected in 7 of the CF group (16%). Parainfluenza 3 was detected in BALF from one of the CF group.
Cell counts, cytokine and protease concentrations in BALF are shown in table 1. Total cell, neutrophil, IL-8 and TGF-β1 concentration were significantly higher in the CF group than in the CRS and control groups; concentrations were similar in the PCD group. With protease inhibition, IL-10 was detected in 29% of CF samples, compared with 83% of PCD, 79% of CRS and 100% of control samples (χ2 = 21.8, p<0.001). Without protease inhibition, IL-10 was detected in 9% of CF samples (p<0.05).
NE activity, MMP-9 concentration and molar ratios of MMP-9 to its inhibitor TIMP-1 were significantly higher in the CF group than in the CRS and control groups but similar to the PCD group, as shown in table 1. TIMP-1 concentrations were similar between groups. In the CF group, NE activity correlated negatively with FEV1 (r = −0.44, p<0.05). MMP-9 concentration did not correlate with any spirometric index, but the molar ratio of MMP-9:TIMP-1 correlated negatively with FEV1 in the CF group (r = −0.47, p<0.05, fig 1). The MMP-9 concentration correlated with the neutrophil concentration in the CF group (r = 0.71, p<0.001).

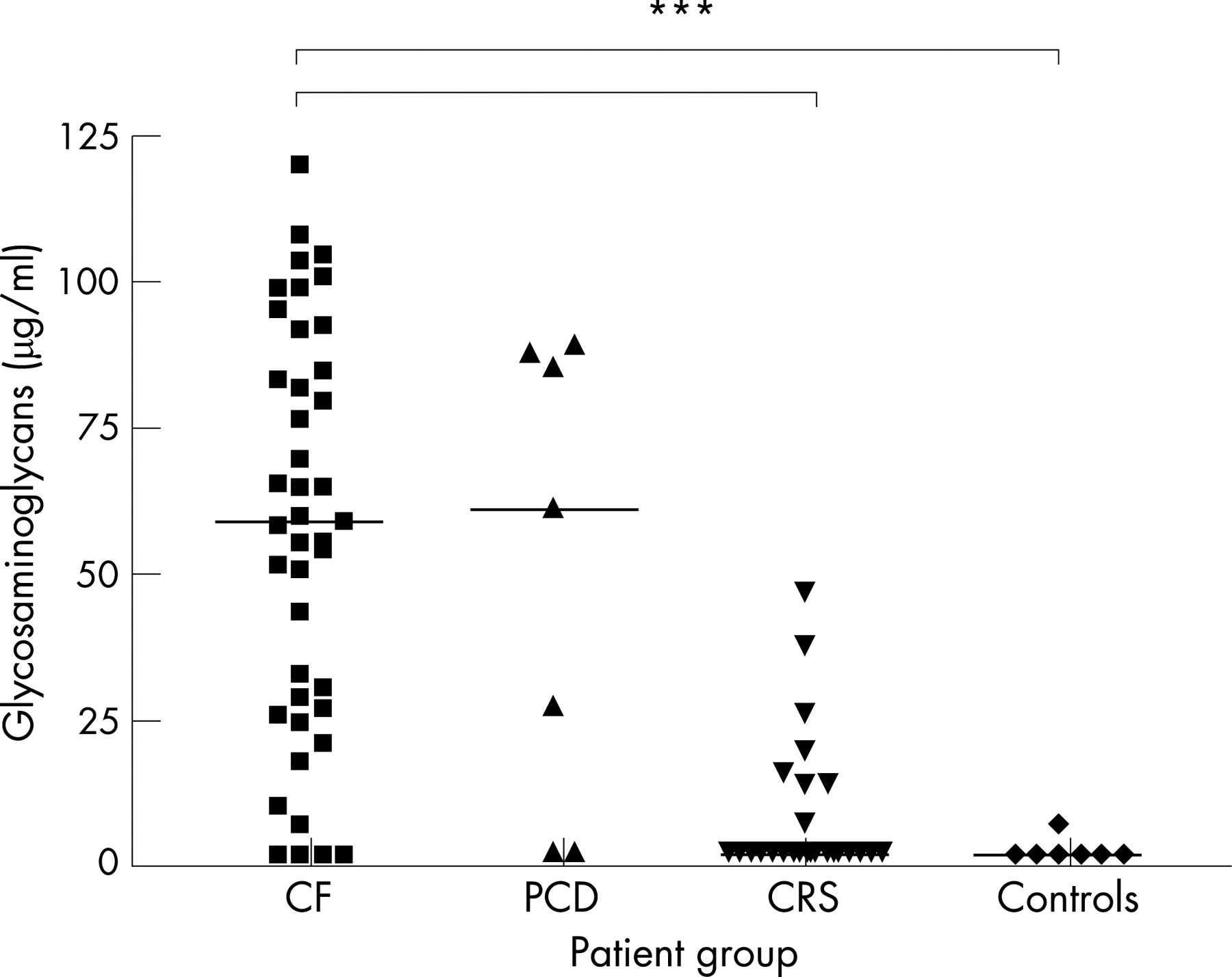
Matrix constituent concentrations are shown in table 1 and individual concentrations of glycosaminoglycans are shown in fig 2. Glycosaminoglycan, elastin and collagen concentrations were significantly higher in the CF group than in the CRS and control groups, but were similar to the PCD group. Within the CF group, correlations of matrix constituent concentrations and age, neutrophil and MMP-9 concentration, NE activity and FEV1 are shown in table 3. In particular, glycosaminoglycans correlated positively with neutrophil concentration (r = 0.85, p<0.001, fig 3) and elastin correlated negatively with FEV1 (r = −0.45, p<0.05, fig 4). In patients with CF under the age of 6 years, 85% had glycosaminoglycan concentrations above the upper limit of the control group, as did 53% for elastin and 62% for collagen.



No marker of inflammation or remodelling correlated with the concentration of pathological bacterial concentration and there was no difference in these parameters between cases with or without Pseudomonas aeruginosa identified in BALF, or between ΔF508 homozygotes and other genotypes The only parameters that were significantly different between CF cases with an exacerbation and those who were well or clinically stable were the total cell count (2.3×106 vs 1.0×106/ml, p<0.05), neutrophil concentration (1.0×106 vs 0.4×106/ml, p<0.05) and glycosaminoglycan concentration (65 vs 27 μg/ml, p<0.05).
RBM thickness
Of the 78 patients in whom endobronchial biopsies were taken, 50 (64%) had at least one biopsy that was of sufficient quality for RBM thickness to be measured. Biopsy success rate was 59% in CF cases and 69% in non-CF cases; there was a lower success rate with small forceps (1.2 mm biopsy channel) compared with large forceps (2 mm biopsy channel) (33% vs 90%; χ2 = 14.54, p<0.001).
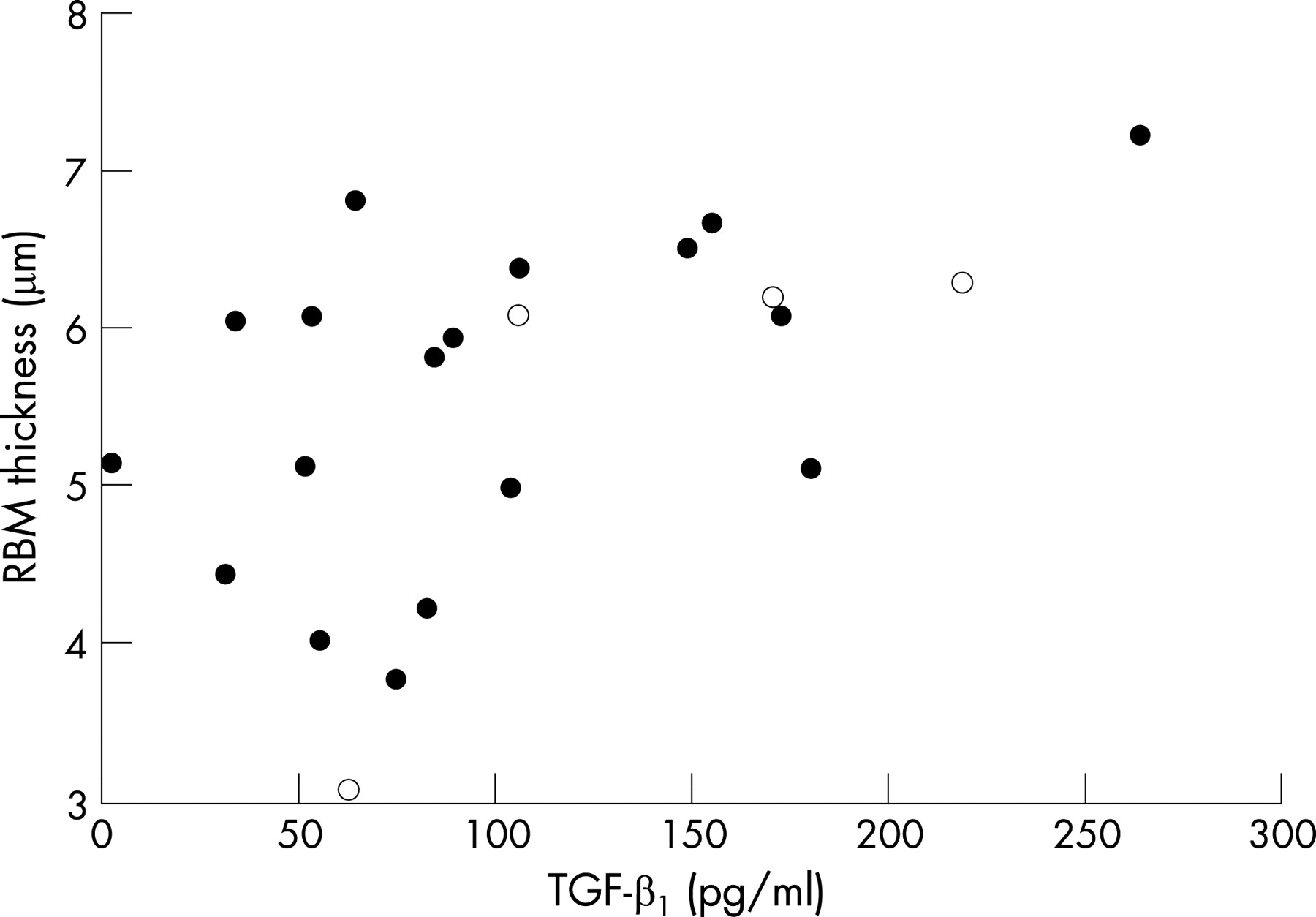
RBM thickness in each patient group is shown in fig 5. Median RBM thickness was significantly greater in the CF group (5.9 μm) than in controls (4.0 μm, p<0.01) but was similar to the CRS (4.6 μm) and PCD groups (4.6 μm). Eighteen of the 24 biopsies (75%) in the CF group had a RBM thickness greater than the upper limit of the control group (4.5 μm). There was no difference in RBM thickness between the 5 patients with CF who were stable and the 19 with an exacerbation (5.8 vs 6.1 μm). In the CF group there was no relationship between RBM thickness and age; RBM of >4.5 μm was present in 5 children with CF under the age of 6 years (including 2 infants). RBM thickness correlated with total TGF-β1 concentration (r = 0.53, p = 0.01, fig 6) but not with pulmonary function tests, dose of inhaled corticosteroids, BALF cell count, pathological bacterial load or other cytokine or protease concentration.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
This is the first study to investigate airway remodelling using both BALF and endobronchial biopsies in a large group of children with CF. We found evidence for two forms of airway remodelling in children with CF; first, matrix breakdown, as demonstrated by increased levels of matrix components in BALF, related to increased neutrophils and proteases and decreased pulmonary function; and second, RBM thickening which was related to TGF-β1 concentration but not to other inflammatory markers or lung function.
This study confirms higher levels of NE and MMP-9 in BALF in CF.9 However, contrary to previous work,9 the concentration of TIMP-1, the inhibitor of MMP-9, was not increased in our patients. In asthma the molar ratio of MMP-9 to TIMP-1 has been found to be lower than in controls and to correlate positively with FEV1,20 with an excess of TIMP-1 implicated in increased collagen deposition.21 In the present study the molar ratio of MMP-9 to TIMP-1 was higher in the CF group than in controls and correlated negatively with FEV1. In asthma, higher levels of TIMP-1 to MMP-9 may protect against airway wall destruction. We speculate that the opposite may be true in CF, where an imbalance of MMP-9 over its inhibitor may result in destruction of the airway wall.
The extracellular matrix is a complex network within the airway wall consisting of fibrous (collagen and elastin) and adhesive (fibronectin and laminin) proteins embedded in a hydrated polysaccharide gel containing glycosaminoglycans which provide rigidity to the airway wall.2 Previous studies have found increased breakdown products of elastin11 22 and collagen11 in the urine of patients with CF, but no clear relationship between these and lung function. Although alteration to the extracellular matrix is a recognised finding in endobronchial biopsies in patients with asthma,23 this is the first study to our knowledge to measure matrix components in BALF. We have demonstrated increased concentrations of elastin, glycosaminoglycans and collagen in BALF in the CF group compared with the CRS and control groups, correlating with neutrophil concentration, NE activity and MMP-9 concentration. This is compatible with the hypothesis that these components are being broken down by proteases and being released into the airway lumen. Elastin concentration also correlated negatively with FEV1, and the glycosaminoglycans concentration was significantly higher in CF cases with an exacerbation. In addition, concentrations of matrix components were increased above control values even in very young children with CF, suggesting that this process begins early. The extracellular matrix appears to be involved in airway development and repair24 and may have a role in cell chemoattraction.25 It is plausible that the presence of matrix components in the airway lumen may themselves act to further heighten inflammation. This could be usefully tested in an in vitro model of neutrophil migration.
Median RBM thickness in the CF group was greater than in the control group but was similar to that in the PCD and CRS groups. The median thickness (6.1 μm) was less than that reported in children with difficult asthma (8.2 μm),19 although similar to that reported for children with mild to moderate asthma.26 RBM thickness in the control cases was similar to that previously reported.26 Although RBM thickness has not been previously measured in CF, biopsy studies have examined the subepithelial area, reporting a fibrotic layer beneath the true basement membrane6 and showing a thickened RBM in one publication.7 RBM thickening would therefore appear to be present in CF and occurs early, contrary to the lack of RBM thickening in infants with wheeze less than 2 years of age.27 There appeared to be a clustering of RBM thickness around 3–4 μm in the CRS group, suggesting that the cases with thicker RBM may be outliers and reflecting the mixed pattern of respiratory disease within this group. RBM thickening would therefore not appear to be a specific feature of asthma, and it has also been reported in other disease states such as atopy.26
In the present study RBM thickness correlated with the TGF-β1 concentration in BALF. Although RBM thickness has been reported to be correlated with TGF-β1 reactivity in adult asthmatic biopsies,28 this has not been previously found with TGF-β1 levels in BALF.29 The correlation with RBM thickness fits well with the known effect of TGF-β1 as a potent in vitro stimulator of collagen production,8 which probably forms part of the RBM. An association has also been found between TGF-β1 polymorphisms and disease severity in CF.30 The numbers in our study were too small to look for an effect of TGF-β1 polymorphisms. However, TGF-β1 immunoreactivity in biopsies from patients with CF has previously been associated with a better FEV1 and fewer exacerbations.7 This would suggest some benefit from the presence of TGF-β1, which also has anti-inflammatory properties.8 The functional consequence of RBM thickening is currently unclear, and it is not known whether it is detrimental to airway function or if it may provide some resistance to airway compression. The discrepancy between airway destructive processes and RBM thickening is intriguing and suggests that RBM thickening could conceivably be a protective response, as has been suggested for asthma.31
There are few data available on the inflammatory profile in the lower airways in PCD, although a recent study of sputum from children with PCD reported a median neutrophil differential of 70%.32 The present study shows a very similar picture to CF in terms of neutrophil, IL-8 and protease concentrations, despite the generally milder clinical phenotype seen in PCD. There were also similar concentrations of matrix components, demonstrating that matrix breakdown is not specific to CF. The high matrix concentrations in BALF from patients with PCD tended to be present in those samples with higher inflammatory markers, so it would be appear to be linked to heightened inflammation as was observed for CF. However, all of this small group were studied during an exacerbation, and this may not therefore be representative of patients with PCD in general.
We also measured concentrations of IL-10, an anti-inflammatory cytokine, and IL-13, a Th2 cytokine which appears to be involved in the development of murine asthma models.33 With the addition of protease inhibitors to thawing BALF, IL-10 was detected in significantly fewer BALF samples in the CF group than in the CRS and control groups, contrary to the findings in previous studies.34 Without the use of protease inhibition, IL-10 was detected in even fewer CF samples which indicates that IL-10 is degraded by proteases in BALF. This suggests that protease inhibition is required for its accurate measurement, contrary to that previously reported.35 IL-13 was similar in all the groups studied and we therefore have no evidence that it is present in altered amounts in CF.
This study has a number of limitations. The opportunistic nature of sampling necessitated that the study was cross-sectional and included children with CF with a wide range of age and disease severity. In addition, patients with exacerbations were studied at differing time points in their disease process when their physicians thought that bronchoscopy was clinically indicated. We cannot therefore confirm whether the correlations between markers of remodelling and inflammation are causative or merely associations. The CRS group was a mixed group of children with a variety of disease processes. The control group provided a better comparison for children who were unlikely to have lower airway disease, but it was small, reflecting the difficulty of obtaining control tissue in this type in children. Because the CF group was relatively young, with a median age of 6 years, only half of the group were old enough to perform spirometric tests. This therefore reduces the power of the study to look for correlations with pulmonary function tests. Furthermore, endobronchial biopsies were taken from a subcarina in the right lower lobe in a fourth to fifth generation airway. CF, however, is a disease process that affects airways throughout the bronchial tree and therefore changes in a relatively large airway may not reflect what is occurring in smaller airways, and which may be demonstrated in BALF. We also have not been able to separate any differential effects of disease between airway or parenchymal components; a limitation of BAL sampling is that it will also reflect changes peripherally in the lung parenchyma. We were not able to obtain biopsy specimens from all children as it was difficult to biopsy the airways using small forceps, especially if there was a lot of mucus in the airway. We would have liked to examine deeper structures in the airway wall but, for obvious reasons, a full thickness biopsy could not be obtained. Although detailed quantitative microbiology was performed on BALF, we did not widen our analysis to historical BALF or non-invasive cultures and we accept that this is a weakness of the study. Our study focused on only two aspects of remodelling—namely, thickness of the RBM and matrix degradation. Further work will examine other features of remodelling such as quantification of airway smooth muscle and matrix components in the submucosa. Combining invasive investigations with pulmonary function tests appropriate for younger children, such as multiple breath washout and imaging (particularly high-resolution CT scanning) may be useful. Finally, this was by definition an exploratory observational study and we had no prior data with which to perform power calculations. We have attempted to limit type 1 error by applying lower p values to analyses involving multiple groups but, as we analysed many outcome variables, our positive results must be interpreted with some caution and ideally reproduced in a second cohort.
In conclusion, this study provides evidence for two forms of airway remodelling in children with CF. Matrix breakdown correlates positively with BALF proteases and negatively with pulmonary function. RBM thickening also occurs in CF and is related to TGF-β1 concentration but not to other inflammatory markers or lung function. These processes occur early in life in CF, and matrix breakdown in particular may be a therapeutic target to delay deterioration in pulmonary function.
Acknowledgments
The authors thank Neil Madden for help with the quantitative microbiology; Chloe Dunn and Bernie Ortega for their assistance with bronchoscopies; Pat Haslam for help with cytospin analysis; the Department of Pathology, Royal Brompton Hospital for their preparation of biopsy material; Ian Balfour-Lynn and Mark Rosenthal for the inclusion of their patients in the study and for performing bronchoscopy. They also gratefully acknowledge the patients and families who agreed to take part in the study.
REFERENCES
Footnotes
This work was supported by a Swiss National Foundation grant 1172/05 and ERS fellowship 64/05 to NR.
Competing interests: None declared.
- Abbreviations:
- BALF
- bronchoalveolar lavage fluid
- CF
- cystic fibrosis
- CFTR
- cystic fibrosis transmembrane regulator
- CRS
- chronic respiratory symptoms
- FEV1
- forced expiratory volume in 1 s
- IL
- interleukin
- MMP
- matrix metalloproteinase
- NE
- neutrophil elastase
- PCD
- primary ciliary dyskinesia
- RBM
- reticular basement membrane
- TGF-β1
- transforming growth factor-β1
- TIMP-1
- tissue inhibitor of metalloproteinases-1