Article Text

Abstract
BACKGROUND Primary pulmonary hypertension (PPH), resulting from occlusion of small pulmonary arteries, is a devastating condition. Mutations of the bone morphogenetic protein receptor type II gene (BMPR2), a component of the transforming growth factor beta (TGF-β) family which plays a key role in cell growth, have recently been identified as causing familial PPH. We have searched for BMPR2 gene mutations in sporadic PPH patients to determine whether the same genetic defect underlies the more common form of the disorder.
METHODS We investigated 50 unrelated patients, with a clinical diagnosis of PPH and no identifiable family history of pulmonary hypertension, by direct sequencing of the entire coding region and intron/exon boundaries of the BMPR2 gene. DNA from available parent pairs (n=5) was used to assess the occurrence of spontaneous (de novo) mutations contributing to sporadic PPH.
RESULTS We found a total of 11 different heterozygous germline mutations of theBMPR2 gene in 13 of the 50 PPH patients studied, including missense (n=3), nonsense (n=3), and frameshift (n=5) mutations each predicted to alter the cell signalling response to specific ligands. Parental analysis showed three occurrences of paternal transmission and two of de novo mutation of theBMPR2 gene in sporadic PPH.
CONCLUSION The sporadic form of PPH is associated with germline mutations of the gene encoding the receptor protein BMPR-II in at least 26% of cases. A molecular classification of PPH, based upon the presence or absence ofBMPR2 mutations, has important implications for patient management and screening of relatives.
- primary pulmonary hypertension
- BMPR2
- BMPR-II
- TGF-β
Statistics from Altmetric.com
Primary pulmonary hypertension (PPH) is characterised by a sustained increase in mean pulmonary artery pressure (>25 mm Hg at rest and >30 mm Hg with exercise at catheterisation) and with no identifiable cause, such as recurrent thromboembolism, chronic hypoxic lung disease, or left sided cardiac disease.1 PPH is twice as common in females than males and symptoms develop typically in the third and fourth decades of life, although the disease may occur at any age.2 The mainstay of treatment is oral calcium antagonists and anticoagulants; however, substantial improvements have been made in treatment during the past decade with the use of continuous intravenous epoprostenol.3 4 When medical therapy is ineffective or unavailable, then heart/lung transplantation is currently the best alternative therapy.4 Until recently, the cellular and molecular basis of this devastating disease has remained elusive.
At least 6% of subjects diagnosed with PPH have a known family history of the disorder.5 Familial PPH (FPPH) segregates as an autosomal dominant trait but with markedly reduced disease gene penetrance.6 Recognition of the inherited form of this disorder enabled linkage, with no evidence of locus heterogeneity, and, most recently, positional cloning of a gene mapped to a locus designated PPH1 on chromosome 2q33.7-11 Mutations in the bone morphogenetic protein receptor type II gene (BMPR2), a receptor member of the transforming growth factor beta (TGF-β) family, have recently been identified as causing FPPH.11
Members of the TGF-β family signal by binding to types I and II cell surface receptors and activating serine/threonine kinases by formation of heteromeric complexes.12 Bone morphogenetic proteins (BMPs) were originally identified as proteins regulating growth and differentiation of bone and cartilage, but more recent studies have shown that these multifunctional cytokines regulate growth, differentiation, and apoptosis of various cell types.12 In both familial and sporadic PPH patients, histological examination of pulmonary arteries shows increased muscularisation of the pulmonary arteries, intimal hyperplasia, endothelial cell proliferation, and in situ thrombus.13 14 These findings, together with the similar clinical features and progression of the disease and the determination of previously unknown co-ancestry between two sporadic PPH patients, suggest that a proportion of sporadic PPH patients may have the same underlying molecular genetic defect as seen in FPPH.15 As resolution of this question has important implications for patient diagnosis, genetic counselling, and clinical screening, we have investigated the BMPR2gene in a cohort of patients with no family history of the disease.
Methods
PATIENTS
Fifty sporadic PPH patients (age range 7-61 years) were recruited through physicians at specialist pulmonary vascular clinics in the UK (n=35), France (n=12), and the USA (n=3). PPH was defined by standard clinical methods, including cardiac catheterisation showing pulmonary hypertension (mean pulmonary artery pressure >25 mm Hg) and a normal pulmonary artery wedge pressure.2 Secondary causes of pulmonary hypertension were excluded including other heart and lung diseases, pulmonary embolism, connective tissue diseases, and the use of appetite suppressants (http://www.who.int/ncd/cvd/pph.html). All studies were performed with consent and approval by the Leicestershire Health Authority Ethics Committee, Royal Brompton & Harefield NHS Trust Ethics Committee, South Manchester Local Research Ethics Committee, West Glasgow Hospitals University NHS Trust, Vanderbilt University IRB, and University of Utah IRB.
DNA SEQUENCE ANALYSIS OFBMPR2 GENE
We obtained 10 to 20 ml of peripheral blood from each person studied. DNA was isolated from whole blood as described elsewhere.8 Parental relationships were confirmed through the segregation analysis of 10 independent highly polymorphic short tandem repeat markers. Protein coding sequences from exons 1 to 13 were amplified from genomic DNA using primers derived from intron sequence as previously described.11 Genomic fragments amplified by the polymerase chain reaction (PCR) were sequenced with a dye terminator cycle sequence system (ABI 3700, Perkin-Elmer Applied Biosystems, Foster City, CA).
CONFIRMATION OF GENOTYPES AND DETECTION OF SPONTANEOUS (DE NOVO) MUTATIONS
Variants of the BMPR2 gene were identified by sequence analysis and, when possible, were independently confirmed by restriction endonuclease digestion. Relevant exons were PCR amplified using primers as described, digested with restriction enzymes (HaeIII,TaqI, MseI,Fnu4HI, New England Biolabs) according to the manufacturer's instructions, and size separated on a 4% composite agarose gel (FMC BioProducts, Gibco BRL). The presence or absence of the sequence variants from available family members and at least 150 normal control chromosomes was determined by analysis of the restriction digest or direct sequencing results.
Results
ANALYSIS OF THE BMPR2GENE
Sequencing of genomic DNA of the panel of sporadic PPH subjects showed a total of 11 novel heterozygous mutations of theBMPR2 gene in 13 of the 50 patients studied. The clinical features of these 13 patients are described in table 1. For patients 1 and 2, the nucleotide sequence showed substitutions of guanine for adenine in exons 2 and 3 respectively (table 2). These change the sequences of codon 60 from TGC to TAC (patient 1) and codon 117 from TGT to TAT (patient 2), both changing a highly conserved encoded amino acid from cysteine to tyrosine (table 2). As these mutations do not result in either gain or loss of a restriction site, genomic sequencing of parental samples was used to show the presence of the mutation in the fathers of both patients.
Clinical and characteristics of sporadic PPH patients with BMPR2 mutations
Details of BMPR2 mutations identified in sporadic PPH patients
Genomic sequencing of patient 8 identified a substitution of thymine for cytosine in exon 11, changing the sequence of codon 483 from TGT to CGT and the encoded amino acid from cysteine to arginine (table 2). No additional family members were available for study. In patients 6 and 10, deletion of an adenine in exon 9 and a guanine in exon 12 both lead to a change in the coding reading frame and predict premature truncation of the 1038 amino acid protein at codon positions 423 and 803, respectively. Analysis of genomic DNA samples from unaffected parents, either by direct sequencing (patient 6) or restriction enzyme analysis with Fnu4HI (patient 10) of the appropriate exonic PCR fragment, showed the absence of the mutation, confirming these patients had spontaneous mutations of theBMPR2 gene (table 2, fig 1). The possibility of incorrect paternity was excluded by the analysis of informative markers. The mutation observed in patient 10 was also detected in two further sporadic patients, 11 and 12, ascertained independently. No parental samples were available and the possibility of the patients having inherited the mutation from a common ancestor was excluded through the examination of genotypes from microsatellite markers from within and surrounding the BMPR2 gene on chromosome 2.

Restriction fragment analysis and segregation of mutations of the BMPR2 gene. (A) The mutation in patient 5 (R322X), detected by loss of a TaqI restriction site, was also present in the patient's asymptomatic father as indicated by the presence of the 237 base pair (bp) fragment. (B) The mutation in patient 10 (2386delG), detected by loss of a Fnu4H I restriction site producing an additional 114 bp fragment, is a spontaneous (de novo) mutation since it is not present in either parent.
In three additional patients, insertions of nucleotide residues in the genomic sequence occurred. In patient 4, an additional thymine was detected in exon 6 at position 787 (table 2). In patient 7 both a guanine and adenine were inserted at nucleotide position 1247-8 of exon 9, while in patient 9 an adenine was inserted in exon 12 at position 1969 and confirmed through restriction digest analysis withMseI (table 2). Each of the mutations predicts premature truncation of the BMPR-II protein through shifts of the reading frame (table 2). Parental material was not available for analysis for these patients.
For the remaining three patients (3, 5, and 13) the substitution of cytosine for thymine at CpG dinucleotides occurred in exons 6, 8, and 12, respectively (table 2). In all three patients the mutations result in the change of the encoded amino acid arginine CGA to the stop codon TGA. These sequence changes were confirmed by restriction digest analysis of exonic PCR products with eitherHaeIII (exons 6 and 12) orTaqI (exon 8). While parental material was not available for patients 3 and 13, restriction analysis showed the mutation to be of paternal origin in patient 5. None of these sequence changes were detected in the analysis of a panel of at least 150 chromosomes from unrelated normal subjects, indicating that these mutations are not polymorphisms. Mutations of the entire protein coding sequence of the BMPR2 gene were also excluded, by direct sequencing, in the remaining 37 patients diagnosed with sporadic PPH.
Discussion
The World Health Organization maintains the broad classification of PPH into familial and sporadic forms (http://www.who.int/ncd/cvd/pph.html). However, several lines of evidence point to the possibility of a common genetic basis for both the sporadic and familial forms of the disease. Firstly, the disease gene, although inherited as an autosomal dominant trait, acts with low penetrance, so in the absence of detailed genealogical data, familial cases may be easily overlooked.15 Secondly, both forms of the disease share the same histopathological features and follow a similar clinical course.1 The present study provides direct evidence that germline mutations distributed throughout theBMPR2 gene predispose to sporadic PPH in addition to the familial form of the disease (table 1, fig2).11
{kind=link}
{kind=link}
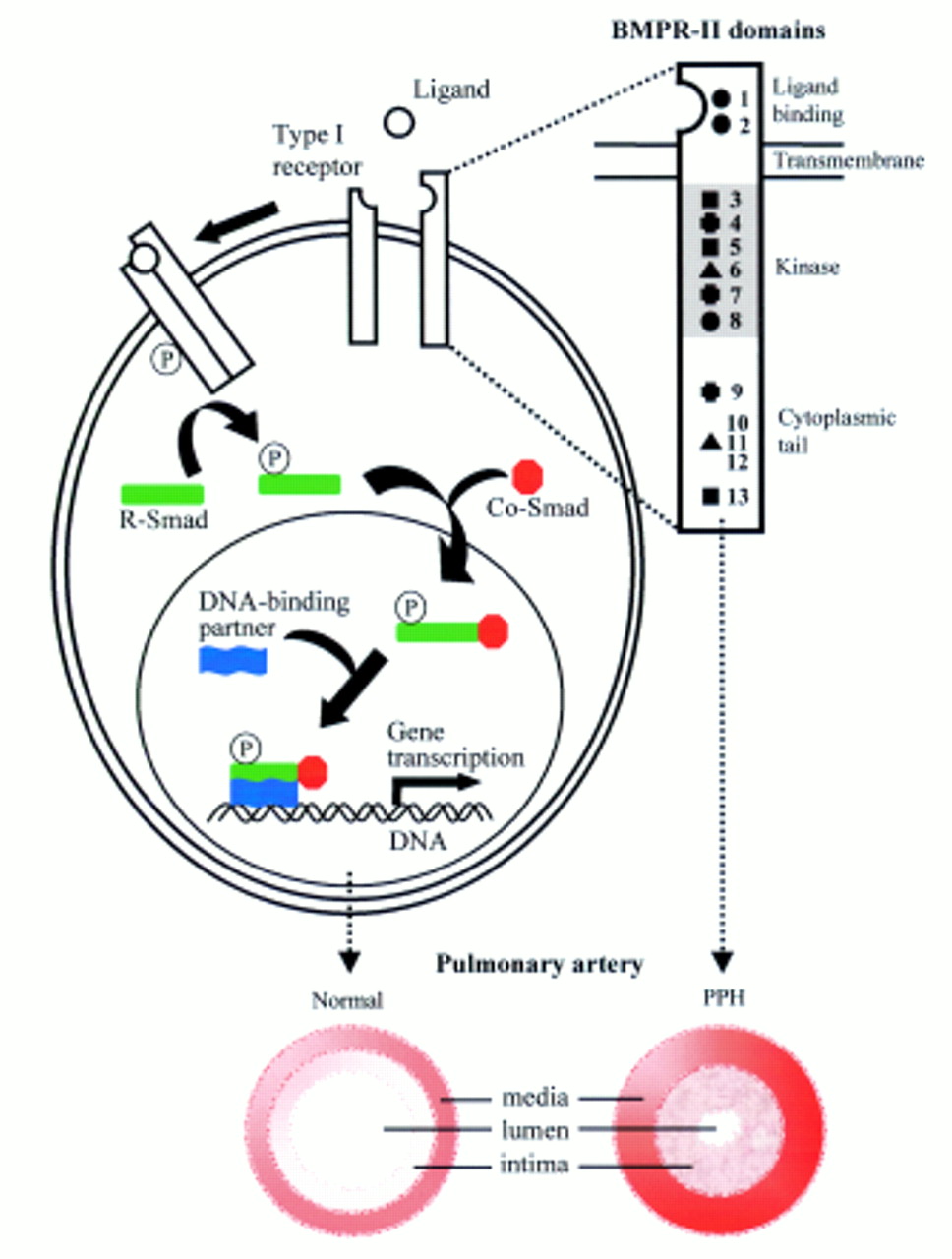
Diagram of the TGF-β signalling pathway and position of BMPR-II mutations. Following ligand binding, BMPR-II forms a heteromeric complex with a type I receptor, resulting in activation of the type I receptor kinase domain. This initiates phosphorylation of cytoplasmic signalling proteins, termed Smads, leading to signal transduction.12 16 The identified heterozygous germline BMPR2 mutations, illustrated on the BMPR2-II molecule (upper right), are likely to perturb the normal cellular effects of pathway activation, including growth inhibition, and extracellular matrix deposition leading to the characteristic histological features seen in the pulmonary vasculature in PPH patients.•=missense mutation,•=nonsense mutation, ✚= insertion, ▴=deletion. The corresponding patient number is noted to the right of the symbol.
We screened the entire protein coding sequence of theBMPR2 gene by direct sequencing and identified heterozygous mutations in approximately one quarter of the study cohort. This strategy, while capable of detecting splice site and coding region mutations, would fail to pick up non-coding region substitutions in either the promoter or introns as well as larger gene deletions and rearrangements. In addition, the direct sequencing methods used in the study may not detect all heterozygous mutations. Hence, the proportion of patients with sporadic PPH harbouring germline BMPR2mutations is likely to be greater than the 26% detected in this study.
To examine the frequency of spontaneous mutation of theBMPR2 gene in patients with sporadic PPH, we looked for the presence or absence of the disease allele in parental samples. DNA was available from both parents for five of the 13 probands studied. In two patients, the mutation was not detected in either parent and had clearly arisen spontaneously (table 2). In each of the remaining three families, paternal transmission was shown yet detailed pedigree analysis failed to indicate any suggestion of preceding disease (table 2). The present study shows germline mutations of BMPR2 to be present in at least 26% of patients without a family history of the disease, yet the familial form of the condition appears to be uncommon.5 A similar range and type of mutation was detected in the sporadic patients as compared to those reported in a recent series of familial PPH patients.11
One possible explanation for the markedly reduced penetrance for this disease gene is that the vascular abnormalities characteristic of PPH are triggered by the acquisition of a second somatic mutation within the BMPR-II signalling pathway. This situation would be analogous to that found in many tumours where somatic mutations in TGF-β growth inhibitory pathways (for example, TGF-βRI, TGF-βRII, and Smad 3 and 4) lead to uncontrolled proliferation.16 In PPH, endothelial cells comprising plexiform lesions show monoclonality, in contrast to a polyclonal origin in secondary pulmonary hypertension.17 Importantly, the only other reported human germline mutation of a type II receptor is a deleterious kinase domain amino acid substitution of TGF-βRII.18 This mutation predisposes to colorectal cancer, with the loss of the wild type allele in the tumour tissue, as predicted by the Knudson “two hit” hypothesis of tumorigenesis. Alternatively, the presence of a heterozygous mutation in the BMPR-II signalling pathway, either constitutional or somatic, together with exposure to an environmental insult could trigger abnormal vascular remodelling. For example, pulmonary hypertension indistinguishable from PPH may present in association with HIV infection or following ingestion of appetite suppressants, most recently of the fenfluramine/dexfenfluramine group.19-21 Additionally, an epidemic of cases was reported following the ingestion of adulterated rapeseed oil in northern Spain.22 Finally, the markedly reduced penetrance of the disorder could arise from the effects of one or more modifier genes, either autosomal or sex linked in nature.
The identification of inactivating heterozygous mutations of theBMPR2 gene in familial and sporadic PPH underlines the importance of the TGF-β super family in the regulation of vascular development and integrity (fig 2). Mutations in the gene encoding endoglin (a TGF-β receptor complex accessory protein) and the putative TGF-β type 1 receptor, ALK-1, have been identified in hereditary haemorrhagic telangiectasia, a condition characterised by arteriovenous malformations.23 The main effects of TGF-β on vascular cells are growth inhibition, cell differentiation, and stimulation of collagen synthesis.12 16 The detailed molecular mechanisms associated with the pathogenesis of PPH will require further investigation, particularly focusing on identifying the key ligands, interacting receptors, and downstream mediated pathways.
PPH is a diagnosis of exclusion and the current absence of specific diagnostic tools frequently contributes to the delay in securing the diagnosis. Molecular genetic analysis, based upon the detection of germline mutations of the BMPR2 gene must now be considered as a potential diagnostic aid for this group of patients. Because the risk for disease in first degree relatives of a newly diagnosed patient with PPH is low, uncertainty has persisted regarding appropriate strategies for clinical screening of at risk family members. It will be of considerable importance to ensure that the use of genetic analysis in the diagnosis of PPH be supported by appropriate genetic counselling, as part of an integrated programme of family based screening. The detection of germline mutations of theBMPR2 gene will enable appropriate targeting of screening and intervention.
Acknowledgments
We wish to express our thanks to the many patients and their parents who provided samples and details of their medical and family histories, and to the Pulmonary Hypertension Association, Silver Spring, MD, USA for their encouragement and support. We acknowledge the many clinicians and colleagues who have provided information for the patients described; in particular we would like to thank Dr Nicholas Morrell and his colleagues for their constructive criticism of this manuscript. This work has financial support from the Medical Research Council (UK), the British Heart Foundation, the LDS Hospital Deseret Foundation, and the National Institutes of Health (HL61997 and HL48164).