Article Text

Abstract
Nonsense mutations that occur more than 50 bases upstream of terminal spliced junctions are generally thought to lead to degradation of the corresponding transcripts by the process of nonsense-mediated mRNA decay. It has also been proposed that some nonsense mutations may affect splicing by the process of nonsense-associated altered splicing (NAS), or by the disruption of a splicing regulatory element. In this study, the effect of the R553X mutation on the splicing of exon 11 of the cystic fibrosis transmembrane conductance regulator gene was investigated. Evidence that R553X causes exon 11 to skip through the creation of a putative exonic splicing silencer (ESS) was provided. The putative ESS appears to be active when located immediately upstream of a 5′ splice site. These findings argue against the possibility that R553X-associated exon 11 skipping is caused by NAS. The study further suggests that aminoglycoside antibiotic treatment would not be effective for patients with the R553X mutation, owing to the skipping of exon 11, and further emphasises the need for detailed mechanistic characterisation of the consequences of nonsense disease mutations.
- CFTR, cystic fibrosis transmembrane conductance regulator
- ESE, exonic splicing enhancer
- ESS, exonic splicing silencer
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- HEK, human embryonic kidney
- IB3, human bronchial epithelial
- NAS, nonsense-associated altered splicing
- NMD, nonsense-mediated mRNA decay
- 2′OMeAO, 2′-O-methyl antisense oligonucleotides
- PTC, premature termination codon
- RT-PCR, reverse transcriptase-polymerase chain reaction
Statistics from Altmetric.com
- CFTR, cystic fibrosis transmembrane conductance regulator
- ESE, exonic splicing enhancer
- ESS, exonic splicing silencer
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- HEK, human embryonic kidney
- IB3, human bronchial epithelial
- NAS, nonsense-associated altered splicing
- NMD, nonsense-mediated mRNA decay
- 2′OMeAO, 2′-O-methyl antisense oligonucleotides
- PTC, premature termination codon
- RT-PCR, reverse transcriptase-polymerase chain reaction
Cystic fibrosis is the most common autosomal recessive disease in the Caucasian population.1 Cystic fibrosis is characterised, in its classic form, by chronic obstructive pulmonary disease, exocrine pancreatic insufficiency, increased sweat chloride electrolytes and male infertility.1 The primary cause is a defect in chloride and sodium transport in cystic fibrosis secretory epithelial cells due to mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The CFTR gene consists of 27 exons that span approximately 200 kb of genomic sequence located on the long arm of human chromosome 7.2 To date, over 1400 CFTR mutations have been identified (http://www.genet.sickkids.on.ca/cftr/). Significant genotype–phenotype correlation has been observed between CFTR mutations and pancreatic function,3 but it has also been recognised that secondary genetic factors modulate the severity of a subset of cystic fibrosis-associated symptoms.4–7
Of the CFTR mutations identified, 10.2% are nonsense mutations and involve nucleotide changes that would apparently result in the creation of premature termination codons (PTCs) (http://www.genet.sickkids.on.ca/cftr/). These mutations are associated with pancreatic insufficiency and a generally severe cystic fibrosis phenotype.8 Transcripts containing nonsense mutations are typically degraded by a quality control mechanism known as nonsense-mediated mRNA decay (NMD) to prevent the synthesis of truncated proteins that may otherwise exert dominant negative effects.9,10 Examples of CFTR nonsense mutations that generate reduced transcript levels include G542X and Y1092X.11 There are also examples of nonsense mutations that cause skipping of the exon in which they reside such as the CFTR R75X11 and R553X mutations.12 Two different mechanisms have been proposed to explain how nonsense mutations cause exon skipping more often than other types of mutations. The first proposed mechanism, known as nonsense-associated altered splicing (NAS), could involve a scanning process that monitors the open reading frame for PTCs before splicing occurs, promoting skipping of the exon in which the PTC is located.13–15 The second proposed mechanism, which is supported by more extensive evidence, involves the inactivation of exonic splicing enhancers (ESEs), which are short and degenerate sequences that promote the selection of proximal splice sites in constitutive and alternative exons.16 It is of interest to note that purine-rich codons (eg, GAA, GGA and AAG) which often overlap strong ESEs17 can be mutated by a single base to the PTCs TAA, TGA and TAG, respectively. Evidence that such mutations disrupt ESE function has been reported.16,18
In addition to ESEs, other splicing elements are known to regulate the inclusion of exons. These include intronic splicing enhancers, and, exonic or intronic splicing silencers, which regulate the selection of splice sites and are often bound by proteins that interfere with the recruitment of the spliceosome to splice sites.19 Two recent studies have identified groups of exonic splicing silencers (ESSs) using bioinformatics20 and systematic evolution of ligands by exponential enrichment21 approaches. The first study identified sequences more frequently present in pseudoexons than in authentic exons and showed that these sequences promoted exon skipping in a reporter system.20 The second study identified sequences from random pools of hexamers and decamers that prevented the inclusion of an exon and were enriched in pseudoexons compared with constitutive exons.21 In follow-up experiments, the mode of action of ESSs was investigated and shown to be position dependent with respect to splice sites.22
Studies have shown that diseases such as cystic fibrosis caused by nonsense mutations may be treated with aminoglycoside antibiotics.23,24 Aminoglycoside antibiotics suppress translational fidelity, resulting in partial expression of the mutated allele. Thus, it is paramount to determine whether stable transcripts are generated from nonsense mutation-containing genes, and to understand the mechanism underlying the disruptive effects of these mutations. To date, distinct results about the effect of the third most common nonsense mutation, R553X, on the CFTR transcript have been reported.12,25,26 In this report, we have carried out a detailed analysis to determine the mechanism by which the CFTR R553X mutation alters gene expression in patients with cystic fibrosis. We provide evidence that the R553X mutation results in the creation of a putative ESS element that acts on 5′ splice site selection leading to exclusion of exon 11.
MATERIALS AND METHODS
The study group consisted of five patients diagnosed with cystic fibrosis based on the presence of clinical symptoms characteristic for the disease.1 All patients were compound heterozygotic for ΔF508 (1652del3, exon 10) and R553X (1789C→T) mutations. Control samples were obtained from healthy individuals and did not carry CFTR mutations. The study was approved by the Human Subjects Review Board of The Hospital for Sick Children.
Total RNA was extracted from cultured lymphoblastoid cells treated with cycloheximide (100 μg/ml in DMSO) or DMSO only for 4 h before harvesting, using RNeasy extraction kit (Qiagen). First strand cDNA was synthesised from 2–3 μg of total RNA by oligo-dT-primed reverse transcription with superscript II reverse transcriptase (Invitrogen) according to the supplier’s directions. A PCR was then performed to generate a fragment spanning exons 10, 11 and 12 of the CFTR transcript using primers X10-5, 5′-GAG GGT AAA ATT AAG CAC AG-3′ and X12-3s, 5′-AGG TAT CCA AAA GGA GAG TC-3′, and, a fragment spanning exons 7 and 8 of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript using primers XGAPD-5, 5′-TGG TAT CGT GGA AGG ACT CA-3′ and XGAPD-3, 5′-GTG TCG CTG TTG AAG TCA GA-3′. Accordingly, the PCR amplification was as follows; 30 s at 94°C, 30 s at 60°C and 30 s at 72°C for 30 cycles using 1.5 units of Taq DNA polymerase (Invitrogen). The PCR products were analysed by electrophoresis on 8% polyacrylamide gel. DNA was then transferred to a nylon membrane (Hybond N+ Pharmacia, Amersham) by Southern blotting. An internal oligonucleotide probe complementary to CFTR exon 10 and GAPDH exon 8 were labelled with the radionucleotide27,32P using terminal deoxynucleotide transferase (Invitrogen) according to the supplier’s conditions. The blots were probed and the bands were visualised by autoradiography. PCR products displaying altered mobility were extracted using MiniElute Gel Extraction Kit (Qiagen) and subjected to automated DNA sequencing analysis.
The minigene reporter systems CD44v5, CD44ΔL and CD44ΔM were described previously.28 Seventeen nucleotides flanking the wild-type and R553X alleles of exon 11 of the CFTR gene were introduced into exon 5 of the CD44 gene by overlapping extension using two rounds of PCR.29 The first round of PCR fragments were amplified using 2 μl of Elongase Enzyme mix (Invitrogen) for each of the three sets of constructs (ΔL3′11WT or R553X; ΔL5′11WT or R553X; ΔM11WT or R553X) using the following primers, CD44-F, 5′-CAT GGA TAA CCA TGT TCA GTC-3′ and ΔL3′WT/R553X-R, 5′-TCT TGC TCg/a TTG ACC TCT TCA TCC TGA TAC TCA TGG-3′, ΔL5′WT/R553X-R or ΔMWT/R553X-R, 5′-TCT TGC TCg/a TTG ACC TCT TCG ATA TCA AGC TCG CGT C-3′; and ΔL3′WT/R553X-F, 5′-GA GGT CAA c/tGA GCA AGA TAC AAG CAC AAG TAA GAA GG-3′, ΔL5′WT/R553X-F, 5′-GAG GTC AAc/t GAG CAA GAC CGG AAC CAC AGC CTC CTT TC-3′ or ΔMWT/R553X-F, 5′-GAG GTC AAc/t GAG CAA GAC GAT GAA GAG GAG ACC CAC-3′ and CD44-R, 5′-TCC ACT AGT TCT AGC TAG AAG-3′. The PCR programme consisted of 20 cycles of 20 s at 94°C, 20 s at 60°C (decreasing the annealing temperature by 0.5°C per cycle) and 1 min at 68°C, followed by an additional 10 cycles of 20 s at 94°C, 20 s at 50°C and 1 min at 68°C. The second round of PCR was performed using the two corresponding fragments for each of the three reporter systems and primers CD44-F and CD44-R with 2 μl of Elongase Enzyme mix (Invitrogen). The PCR programme used for the second round consisted of 20 cycles of 30 s at 94°C, 30 s at 50°C (increasing the annealing temperature 0.5°C per cycle) and 2 min at 68°C, followed by an additional 10 cycles of 30 s at 94°C, 30 s at 60°C and 2 min at 68°C. The resulting PCR fragments of 2490 bp were digested using BglII and HpaI and cloned into the V5CD44ΔL and V5CD44ΔM, respectively, using T4 DNA ligase (Invitrogen). The generated minigene reporters were verified by sequencing.
Human embryonic kidney (HEK) 293 cells (from American Type Culture Collection) were grown in Dulbecco’s modified Eagle’s medium with high glucose supplemented with 10% fetal calf serum and 1% streptomycin–penicillin antibiotics. Human bronchial epithelial (IB3) cells30 were grown in LHC-8 medium with glutamine supplemented with 5% fetal calf serum and 1% streptomycin–penicillin antibiotics. The cells were grown in 10 cm tissue culture dishes.
A total of 4 μg of total plasmid DNA was transfected into HEK 293 and IB3 cells in replicates using lipofectamine reagent (Invitrogen). After 18 h of transfection, the medium was replaced with fresh complete medium. Cells were harvested 48 h after transfection. Total RNA was extracted as described previously. Oligo dT-primed cDNA synthesis was preformed as described previously. PCR amplification (30 s at 94°C, 30 s at 60°C and 30 s at 72°C for 30 cycles using 1.5 U of AmpliTaq DNA polymerase (Boehringer Ingelheim)) of the fragment containing CD44 exon 5 was performed using previously described primers.28 The fragments were separated on a 2% agarose gel. The ethidium bromide-stained band intensities were measured by analysis of digitised images using AlphaEase V.5.1. Statistical analysis was performed using GraphPad Prism 4.
RESULTS
Analysis of CFTR transcripts in patient cells carrying the R553X mutation
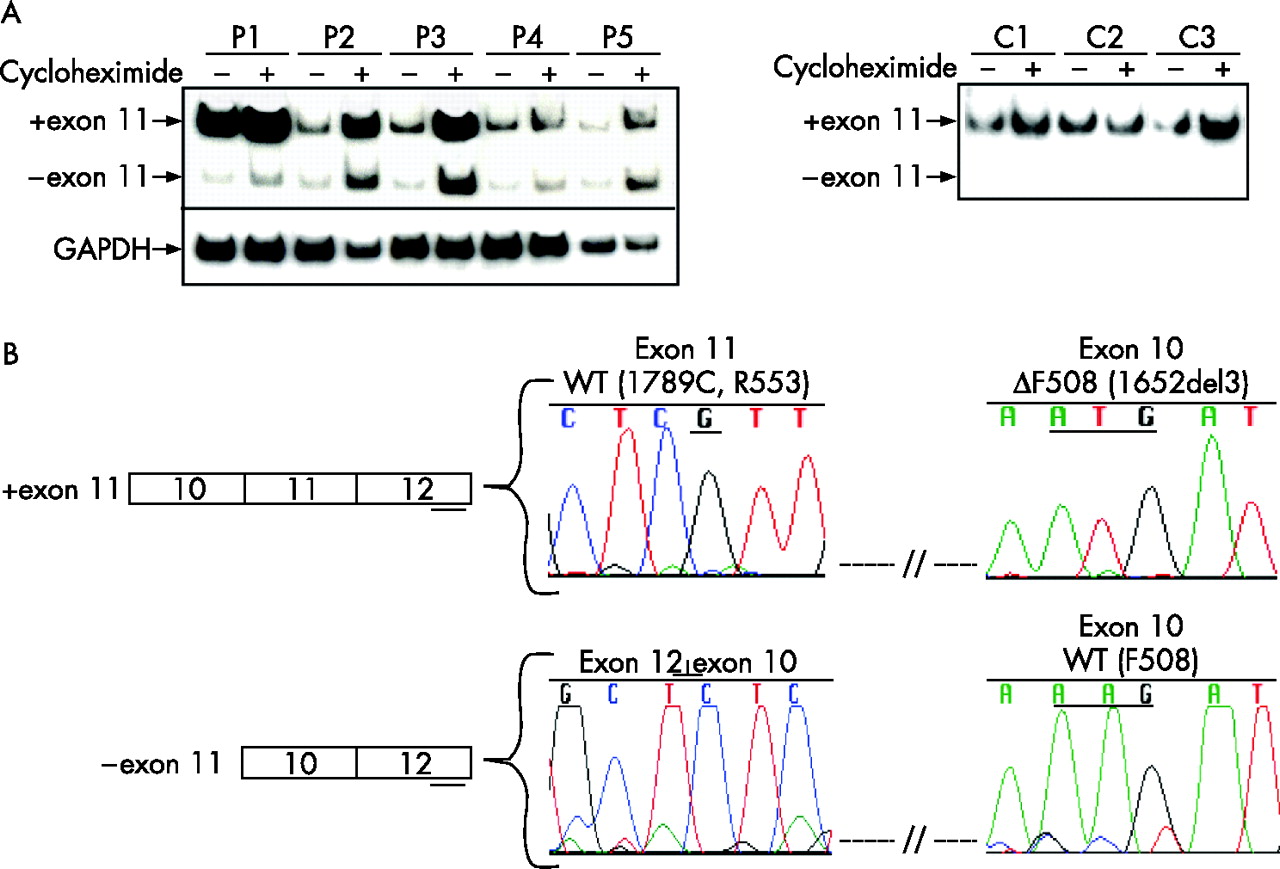
We have performed transcript analysis of five patients with cystic fibrosis who are compound heterozygotic for ΔF508 (1652del3, exon 10) and R553X (1789C→T) mutations. These patient cell lines were used as the ΔF508 transcript has been shown to be stable,31 and can be distinguished from transcripts resulting from the R553X allele. Lymphoblastoid cell lines were available, and we and others previously showed that the splicing patterns of CFTR in these cell lines reflect those observed in airway epithelia.11,32 Accordingly, total RNA was extracted and a fragment spanning CFTR exons 10, 11 and 12 was analysed by reverse transcriptase-polymerase chain reaction (RT-PCR) assays. Variable levels of an aberrantly spliced form that corresponded to the skipping of exon 11 was observed in all five patients with cystic fibrosis that was absent in all control cell lines studied (fig 1A). Two of the three previous studies have shown that no transcript is generated from the R553X allele; however, it should be noted that for one,25 the assay used would not have permitted the detection of the exon 11 transcript and the second,26 did not use a sensitive assay. The results from our study are consistent with the third study12 that also showed that the R553X mutation causes skipping of CFTR exon 11.

Analysis of transcripts from patient cell lines carrying the R553X mutation. (A) Reverse transcriptase-polymerase chain reaction analysis of lymphoblastoid cell lines derived from patients with cystic fibrosis (P1-P5) carrying the ΔF508/R553X genotype and controls (C1–C3) separated by 8% polyacrylamide gel. Cell lines were untreated (–) or treated (+) with cycloheximide (100 μg/ml) for 4 h before harvesting. Lower panel corresponds to endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript. (B) Schematic representation and sequences of the + exon 11 and – exon 11 transcripts. Rectangular boxes denote exons and short lines denote sequencing primers. The chromatographs correspond to the antisense strand sequence of untreated PCR products of patient 2. The upper chromatographs show sequencing results of + exon 11 transcript, indicating the wild-type exon 11 sequence (R553 allele) and the ΔF508 (3 bp-deletion in exon 10) mutation only. The lower chromatographs show that the shorter transcript corresponds to the skipping of exon 11, and that the ΔF508 mutation does not occur in this transcript. P1–P5 = patients 1–5, C1–C3 = controls 1–3, + exon 11 = full-length transcript, − exon 11 = transcript that lacks exon 11.
We assessed whether the skipped transcript (−exon 11) generated from the R553X allele arises from nonsense-associated altered splicing (NAS) and whether it is targeted by the NMD pathway, as the exclusion of exon 11 results in a frameshift that introduces a new PTC in exon 12. For this purpose, the patient with cystic fibrosis and control cell lines was treated with cycloheximide for 4 h before harvesting. Cycloheximide, a protein synthesis inhibitor, has been reported to inhibit NAS27 as well as NMD.33 If the – exon 11 transcript were generated by NAS, a relative reduction of this transcript would be found in the presence of cycloheximide. If there were no NAS, both the + exon 11 and – exon 11 transcripts would be expected to increase in the presence of cycloheximide owing to suppression of NMD. Results of transcript analysis showed an increase in the overall levels for both the + exon 11 and – exon 11 transcripts as compared with endogenous GAPDH transcript levels (fig 1A). Moreover, the expression of the – exon 11 transcript was apparently not reduced on cycloheximide treatment. Although it should be kept in mind that the efficiency with which cycloheximide blocks NAS and NMD processes may vary, these results indicate that NAS is not likely to be the underlying mechanism for exon skipping.
Subsequently, RT-PCR products spanning exons 10–12 from cycloheximide-treated and untreated cell lines derived from patient 2 (fig 1A) were sequenced. Results confirmed that the full-length transcript was generated from the allele carrying the ΔF508 mutation, although the transcript lacking exon 11 resulted exclusively from the R553X allele. In either cycloheximide-treated or untreated cells of patient 2 (fig 1B) the R553X allele was specifically aberrantly spliced. We cannot exclude the possibility that low amounts of the R553X allele may lead to + exon 11 transcript owing to limitations of detection by sequencing analysis. Together, these results suggest that the primary consequence of the R553X mutation is the skipping of CFTR exon 11 and that it is not likely to be due to NAS.
Creation of a putative exonic splicing silencer element by the R553X mutation
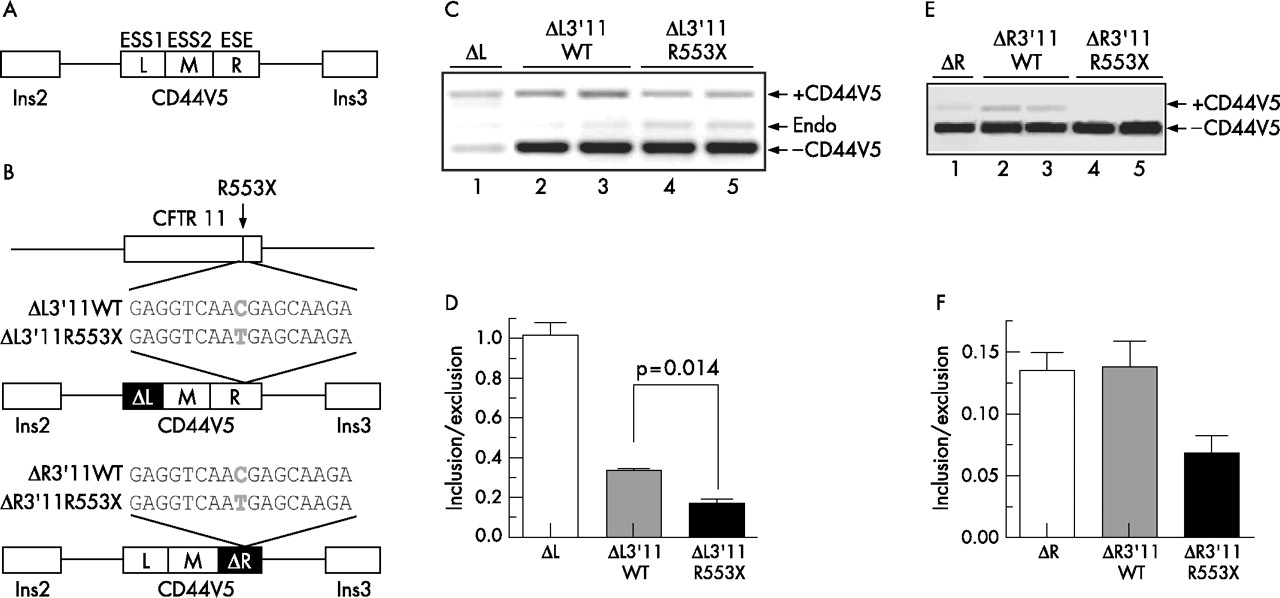
To elucidate more precisely the mechanism underlying R553X mutation-associated exon skipping, a heterologous splicing reporter construct was used. The reporter system was chosen such that it allowed the assessment of sequences that promoted (ESE) or hindered (ESS) exon inclusion. Briefly, it consisted of three exons: two constant exons derived from the human insulin gene that flank the alternatively spliced exon 5 (v5) of the CD44 gene.28 In a previous study,28 two ESSs (L and M) and one ESE (R) were defined by mutagenesis within the CD44 v5 exon (fig 2A). Typically, the intact exon v5 is efficiently skipped, but removal of either of the two ESSs sequences (ΔL and ΔM) increases the level of inclusion such that approximately 50% of the total CD44 reporter transcripts contain the alternatively spliced exon.28,34 Splicing reporter cassettes were generated to assess whether the R553X allele caused exon skipping by disrupting an ESE or by creating an ESS independently of other CFTR transcript features.

Analysis of the consequences of the R553X mutation in a heterologous context indicates that it confers exonic splicing silencer (ESS) activity. (A) Schematic representation of the splicing reporter construct.28 L = ESS1, M = ESS2, R = exonic splicing enhancers (ESE), ins 2 and 3 = insulin exons 2 and 3, CD44V5 = exon v5 of the CD44 gene. (B) Schematic representation of the ΔL and ΔR constructs,28 in which 17 nucleotide sequences flanking the wild-type allele (nt C) or the R553X allele (nt T) were introduced in the 3′ segment of the CD44 exon v5, in the absence of ESS1 (L) or ESE (R) segments. The resulting constructs were named ΔL3′11WT and ΔR3′11WT or ΔL3′11R553X and ΔR3′11R553X, respectively. The blackened ΔL or ΔR boxes denote the substitution of each of these fragments of the CD44 exon v5 with BlueScript sequences as described elsewhere.28 (C) Reverse transcriptase-polymerase chain reaction (RT-PCR) analyses of human embryonic kidney (HEK) 293 cell lines transfected with reporter constructs ΔL (lane 1), ΔL3′11WT (lanes 2 and 3) and ΔL3′11R553X (lanes 4 and 5), respectively. A representative gel of two independent transfection experiments is shown. (D) Measurement of the resulting PCR bands was carried out using AlphaEase V.5.1. The ratio of the band intensity corresponding to the +CD44V5 transcript over the band intensity corresponding to the -CD44V5 transcript (inclusion/exclusion) within each lane was calculated. The bars correspond to the means and standard error calculated for the ratios from four independent transfection experiments. The white bar corresponds to ΔL, the light grey bar corresponds to ΔL3′11WT, and the black bar corresponds to ΔL3′11R553X transcripts. Mann–Whitney U test was performed using GraphPad Prism 4 indicating a statistically significant increase in the skipping of CD44 exon 5 in the presence of cystic fibrosis transmembrane conductance regulator (CFTR) sequences carrying the R553X allele. Similar results were obtained from transfection experiments using the human bronchial epithelial (IB3) cell line (data not shown). (E) RT-PCR analyses of HEK 293 cell lines transfected with reporter constructs ΔR (lane 1), ΔR3′11WT (lanes 2 and 3) and ΔR3′11R553X (lanes 4 and 5), respectively. (F) Measurement of the resulting PCR bands was carried out as described in D. The bars correspond to the means and standard error calculated for the ratios from two independent transfection experiments. The white bar corresponds to ΔR, the light grey bar corresponds to ΔR3′11WT, and the black bar corresponds to ΔR3′11R553X transcripts. Results show an increase in the skipping of CD44 V5 in the presence of CFTR sequences carrying the R553X allele. All PCR products in C and E were separated in a 2% agarose gel. + CD44V5, full-length transcript; −CD44V5, transcript lacking CD44 V5 exon; endo, endogenous insulin transcript.
A region comprising 17 nucleotides surrounding the wild-type (GAG GTC AAC GAG CAA GA) and R553X (GAG GTC AAT GAG CAA GA) alleles of CFTR exon 11 was introduced into the 3′ region of the CD44 reporter exon that lacked the first ESS element (ΔL). This construct maintained the position of the R553X mutation with respect to its distance from the 5′ splice site in CFTR exon 11 (fig 2B). The resulting constructs ΔL3′11WT and ΔL3′11R553X in addition to the parental construct (ΔL) were transfected into HEK 293 and IB3 cell lines. Analyses of transcripts showed that there is a relative increase in exon v5 skipping for ΔL3′11R553X transcripts, compared with ΔL3′11WT transcripts (fig 2C, compare lanes 2 and 3 with 4 and 5). The ratios of exon inclusion/exclusion from the wild-type and mutant constructs were found to be significantly different (n = 4, p = 0.014; Mann–Whitney U test; fig 2D), suggesting that the sequence carrying the R553X mutation may act as a silencer element. It is interesting to note, however, that both constructs containing CFTR sequences (wild type and R553X) showed a dramatic increase in exon skipping compared with the parental construct (ΔL; fig 2D). The 17-nucleotide wild-type sequence may act as a negative element on the splicing of the reporter exon, although the observed effect probably reflects the disruption of the ESE (R) by the CFTR sequences (fig 2B).
To evaluate these possibilities, we constructed additional reporter cassettes in which the ESE (R) was specifically deleted,28 maintaining the position of the CFTR wild-type or the R553X mutation-containing sequences with respect to the 3′ end of the exon (ΔR3′11WT or ΔR3′11R553X, respectively, fig 2B). These constructs together with the parental construct (ΔR) were transfected into HEK 293 cells. The results suggest that the observed increase in exon skipping resulting from the ΔL3′11WT construct with respect to the parental ΔL construct (fig 2D) was due to the disruption of the ESE (R) since only the construct ΔR3′11R553X showed an increase in exon skipping relative to the parental construct (ΔR; fig 2E,F).
Together, these findings suggest that the R553X mutation may result in the creation of an ESS element. Interestingly, the sequence created by the R553X mutation (TCA AGG → TCA ATG) partially resembles the consensus sequence of the ESS “PS10” (A/GAAATG) identified by Zhang and Chasin.20 In fact, their analysis of a representative sequence of PS10 ESS by mutagenesis (TAGAAATG → TAGAAAAG) showed a reduction in ESS activity.20
Positional effect of the R553X mutation on exon skipping
Given the proximity of the splicing element created by the R553X mutation to the 3′ end of exon 11, we hypothesised that this putative negative element may affect 5′ splice site selection. To assess this, we introduced 17 nucleotides of exon 11 flanking the wild-type (GAG GTC AAC GAG CAA GA) and R553X (GAG GTC AAT GAG CAA GA) alleles into the 5′ end (ΔL5′11WT or R553X) and middle (ΔM11WT or R553X) exon v5 segments (fig 3A). These reporter constructs, in addition to the parental constructs ΔL and ΔM, were transfected into HEK 293 cells. Transcript analyses showed no marked changes in exon skipping resulting from constructs carrying the wild-type or R553X CFTR sequences near the 5′ end or middle of the exon (fig 3B,C). Moreover, no significant difference was observed between the constructs carrying the CFTR sequences and either parental constructs (ΔL and ΔM; fig 3B,C). These results suggest that the element created by the R553X mutation does not function as an ESS in the context of a 3′ splice site and may have a negative effect only on a proximal 5′ splice site.

{kind=link}
{kind=link}
{kind=link}
Position effect of the R553X mutation. (A) Schematic representation of the CD44V5 ΔL and CD44V5 ΔM constructs described elsewhere,28 in which 17 nucleotide sequences flanking the wild-type allele (nt C) or the R553X allele (nt T) were introduced at the 5′ and in the middle segments of CD44 exon v5. The resulting constructs were named ΔL5′11WT and ΔM 11WT or ΔL5′11R553X and ΔM11R553X, respectively. ins 2 and 3 = insulin exons 2 and 3, CD44V5 = exon 5 of the CD44 gene. ΔL or ΔM in black boxes denote the substitution of the L (exonic splicing silencer (ESS)1) or M (ESS2) segments of the CD44 exon v5, respectively, with BlueScript sequences as described elsewhere.28 (B) Reverse transcriptase-polymerase chain reaction (RT-PCR) analysis of human embryonic kidney (HEK) 293 cell lines transfected with reporter constructs ΔL (lane 1), ΔL5′11WT (lane 2), ΔL5′11R553X (lane 3), ΔM (lane 4), ΔM11WT (lane 5) and ΔM11R553X (lane 6), respectively. PCR products were separated in a 2% agarose gel. + CD44V5 = full-length transcript, −CD44V5 = transcript lacking CD44 exon 5. (C) Measurement of the resulting PCR bands was carried out as described in fig 2D. The bars correspond to the means and standard error calculated for the ratios from two independent transfection experiments. The white bar corresponds to the ΔL or Δ M, the light grey bars correspond to ΔL5′11WT and ΔM11WT and the black bars correspond to ΔL5′11R553X and ΔM11R553X transcripts.
DISCUSSION
Skipping of exon 11 caused by the R553X mutation through the creation of a position-dependent ESS
To date, more than 146 nonsense mutations identified in the CFTR gene have been associated with severe cystic fibrosis disease presentation (http://www.genet.sickkids.on.ca/cftr/). Studies have shown that PTCs elicit nonsense-mediated mRNA decay, a quality control mechanism that prevents the expression of truncated proteins.9,10 There are examples, however, where nonsense mutations are associated with exon skipping.18 In the present study, we have investigated the CFTR R553X mutation at the transcript level. Exon 11 skipping in patients with cystic fibrosis that are compound heterozygotic for ΔF508/R553X mutation was specifically associated with the R553X mutation. The extent of exon skipping was variable but maintained on cycloheximide treatment. These observations exclude NAS as the cause for R553X-associated exon skipping.
We further investigated the underlying cause of the R553X mutation-associated exon 11 skipping and showed that 17 nucleotides of exon 11 carrying the R553X mutation had a negative effect on splicing in a heterologous reporter system (CD44 v5 exon), compared with the wild-type allele. This effect was only apparent when the R553X mutation was positioned proximal to the 3′ end of the exon, as occurs in CFTR exon 11. The wild-type allele did not have a positive effect on the splicing of CD44 exon 5, indicating that the R553X mutation did not affect an ESE. Thus far, previous studies have shown that nonsense mutations could affect splicing by disrupting ESEs.16,18 Other reports have implicated a quality control mechanism known as nonsense-associated altered splicing13,15 as the underlying cause of nonsense mutation-related exon skipping. Our study provides the first evidence for a nonsense mutation creating an ESS, and further shows that this putative ESS element may result in exon skipping by preventing selection of the proximal 5′ splice site. Interestingly, the sequence created by the R553X mutation partially resembles the consensus sequence of an ESS previously identified by Zhang and Chasin.20 A recent study has shown that the mode of action of ESSs results in the silencing of 5′ splice sites that are located 3′ of the ESS sequences.22 Although Wang et al22 also showed that several such sequences resulted in the silencing of 3′ splice sites located 5′ of the ESSs (in addition to the silencing of the 5′ splice sites), one of them (hexamer “A”, fig 1D22) only influenced the 5′ splice site.22 The putative ESS sequence created by the R553X mutation seems to represent an additional example of this latter mode of action of ESSs.
Therapeutic implications
Our findings have implications for proposed treatments for nonsense mutations involving aminoglycoside antibiotics that were shown to promote read-though of PTCs.23,24 Clinical trials using gentamicin administered to patients with cystic fibrosis carrying the W1282X mutation showed improvement of nasal potential difference measurements and increased CFTR at the cell membrane, compared with patients with cystic fibrosis with non-nonsense mutations.24 However, our findings suggest that nonsense mutations, such as CFTR R553X, which cause exon skipping or are targeted by NMD, may not be good candidates for such treatment. The results thus emphasise the importance of determining the specific consequences of nonsense mutations at the transcript level.
Molecular treatments directed against splicing defects are being tested using various methods such as modified RNA oligonucleotides,35 molecular chimeras,36 splicing factors37 and chemical compounds.38 It was recently shown that 2′-O-methyl antisense oligonucleotides (2′OMeAOs) directed against the Duchenne muscular dystrophy gene systemically delivered to the dystrophic mdx mouse caused the skipping of an exon carrying a nonsense mutation, leading to an improvement in muscle function.35 These results suggest that 2′OMeAOs could also be used to treat patients with splicing defects. Similarly, it may be possible to treat the splicing defect caused by the R553X mutation using a 2′OMeAO directed against the region where the mutation is located in CFTR exon 11, to prevent the binding of splicing factors responsible for exon 11 skipping. The correction would lead to the presence of CFTR transcripts carrying exon 11 generated from the R553X allele, and provided it is not entirely degraded by NMD, it could then be treated using aminoglycoside antibiotics.
Key points
-
The third most common nonsense mutation in the cystic fibrosis transmembrane conductance regulator gene, R553X (1789C→T), was found to cause exon 11 skipping in cell lines from patients with cystic fibrosis.
-
The R553X mutation-associated exon skipping did not seem to result from nonsense-associated altered splicing.
-
The R553X mutation caused exon skipping through the creation of a position-dependent exonic splicing silencer.
Acknowledgments
We thank Dr Andrew Paterson for advice on statistical analyses. The work was supported by a grant from the Canadian Cystic Fibrosis Foundation (CCFF) to L-CT and JZ. IA was the recipient of CCFF Studentship and The Hospital for Sick Children research training awards. BJB was supported by grants from the Canadian Institutes of Health Research.
REFERENCES
Footnotes
-
Competing interests: None declared.