Article Text

Abstract
Objectives: To determine the in vitro effects of unfractionated heparin, fractionated heparin and direct thrombin inhibition on platelet–monocyte aggregation, and to establish the in vivo effects of unfractionated heparin and direct thrombin inhibition on platelet–monocyte aggregates in patients scheduled for percutaneous coronary intervention (PCI).
Design: Platelet–monocyte aggregates were assessed in whole blood from 18 healthy volunteers after the addition of unfractionated heparin (1 U/ml), enoxaparin (0.8 U/ml) or lepirudin (5.6 µg/ml), and in 28 patients scheduled for elective PCI before and after administration of 100 U/kg of unfractionated heparin or 0.75 mg/kg bivalirudin. The influence of P-selectin-mediated platelet–monocyte aggregation was assessed with specific blocking antibodies.
Results: Addition of unfractionated heparin in vitro was associated with a higher level of platelet–monocyte aggregates than in controls (20.1 (1.9)% v 16.2 (1.6)%, respectively, p < 0.001). However, platelet–monocyte aggregation was not affected by enoxaparin or lepirudin (16.9 (2.0)% and 17.0 (2.2)%, respectively, NS). Intravenous unfractionated heparin in vivo also resulted in an increase in platelet–monocyte aggregates (absolute Δ 7.1 (2.7)%, p < 0.01), whereas intravenous bivalirudin had no effect (absolute Δ −1.5 (2.4)%, NS). The addition of P-selectin blockade abolished any increase in platelet–monocyte aggregates associated with heparin.
Conclusions: In vitro and in vivo unfractionated heparin is associated with increased platelet–monocyte aggregation through a P-selectin-dependent mechanism. These findings provide a potential explanation for the superior cardiovascular outcomes associated with fractionated heparins and direct thrombin inhibitors.
Statistics from Altmetric.com
Coronary thrombosis has a central role in the pathogenesis of acute coronary syndromes and the complications of percutaneous coronary intervention (PCI).1 Thrombin is both a key component of the coagulation cascade and a potent platelet agonist. Unfractionated heparin has been a cornerstone of antithrombotic treatment for many years. However, unfractionated heparin has important limitations and newer antithrombotic agents, such as the fractionated heparins and the direct thrombin inhibitors, have recently been developed.
Circulating activated platelets bind to leucocytes, predominately monocytes, to form platelet–leucocyte aggregates. Platelet–monocyte aggregates are a sensitive marker of platelet activation and are raised in acute coronary syndromes, after PCI and during coronary artery bypass grafting.2–5 Recently, it has become clear that adhesion of activated platelets to monocytes has important functional consequences. Platelet–monocyte binding induces expression of cytokines, chemokines, adhesion molecules and tissue factor.6,7,8,9,10 Furthermore, platelet–monocyte aggregation promotes monocyte adhesion to activated endothelium and recruitment of monocytes to sites of arterial injury.11,12 Thus, platelet–monocyte aggregation not only is a sensitive marker of platelet activation but also has important proinflammatory and prothrombotic consequences.
The effect of unfractionated heparin and the newer anticoagulant drugs on platelet activation and platelet–monocyte interactions has not been clearly defined. The objectives of this study were to determine the effects of unfractionated heparin, fractionated heparin (enoxaparin) and direct thrombin inhibition (lepirudin) on platelet–monocyte aggregation in vitro, and to investigate the effects of unfractionated heparin and direct thrombin inhibition (bivalirudin) on platelet–monocyte aggregates in vivo.
METHODS
Study population and blood sampling
In vitro healthy volunteer studies
Peripheral venous blood was obtained from 18 healthy volunteers, aged between 20–35 years, who were taking no drugs. Ethical approval was obtained from the local research ethics committee and all participants provided written informed consent. Blood was drawn by clean venepuncture of a large antecubital vein with a 19 gauge needle and anticoagulated with sodium citrate (final concentration 0.106 mmol/l, Sarstedt Monovette). Care was taken to ensure a smooth blood draw and the minimal necessary tourniquet pressure was used. All samples were processed within 5 min of drawing blood. Blood samples from the 18 volunteers were each incubated at room temperature for 15 min with unfractionated heparin (1 U/ml), enoxaparin (0.8 U/ml) or lepirudin (5.6 µg/ml). Platelet–monocyte aggregates were then immunolabelled and assessed as described below.
The in vitro concentrations of anticoagulants were calculated to reflect concentrations of heparin, enoxaparin and lepirudin used in clinical practice. Once we had shown that lepirudin did not affect platelet activation at 5.6 μg/ml or 56 μg/ml (data for 56 μg/ml not shown), lepirudin at the higher concentration was used as baseline anticoagulation for the later studies. Sodium citrate was not used to avoid the associated calcium depletion and therefore allow assessment of platelet monocyte interactions in a more physiologically relevant environment.
To determine whether P-selectin-mediated platelet–monocyte adhesion was affected by unfractionated heparin, blood was collected from eight healthy volunteers and anticoagulated with lepirudin (56 µg/ml). Platelet surface expression of P-selectin and platelet–monocyte aggregates were quantified by two-colour flow cytometric analysis in whole blood alone and after the addition of unfractionated heparin (1 U/ml). In addition, a function-blocking monoclonal antibody specific for CD62P (CLB-thromb/6, final concentration 10 µg/ml) was added to whole blood 5 min before the addition of unfractionated heparin (1 U/ml). After the addition of unfractionated heparin, the samples were incubated at room temperature for a further 15 min before immunolabelling.
In vivo patient study
Blood samples were obtained from 28 patients scheduled for PCI and 14 patients undergoing diagnostic coronary angiography. Patients were aged between 50–80 years and all were treated with aspirin. Exclusion criteria were treatment with an anticoagulant within the preceding month, myocardial infarction within the preceding three months, PCI or surgery within the preceding three months, renal or hepatic impairment, inflammatory disorders or malignancy. Ethical approval was obtained from the local research ethics committee and all patients provided written informed consent.
Blood samples were obtained from the 28 patients scheduled for elective PCI at baseline and 5 min after administration of either 100 U/kg unfractionated heparin (n = 14) or 0.75 mg/kg bivalirudin (n = 14). Bivalirudin was chosen as the direct thrombin inhibitor for the in vivo study, as its safety and efficacy during PCI have been established. Two blood samples, 5 min apart, were also obtained from 14 control subjects undergoing diagnostic coronary angiography. No angiography or instrumentation of the coronary arteries was performed between blood sampling. All samples were immunolabelled within 5 min of sampling. Platelet–monocyte aggregation was determined by two-colour flow cytometric analysis as described below.
Antibodies and other reagents
All chemicals were obtained from Sigma Chemical Company unless otherwise stated. Phycoerythrin-conjugated CD14 (Tuk-4, IgG2a), phycoerythrin-conjugated CD62P (TRAP1, IgG1) and their appropriate isotype controls were obtained from DakoCytomation (Buckinghamshire, UK). Fluorescein isothiocyanate-conjugated CD42a (GRP-P, IgG1) and control IgG1 were obtained from Serotec Ltd (Oxford, UK). Function-blocking CD62P monoclonal antibody (CLB-thromb/6, IgG1) was obtained from CLB (Amsterdam, The Netherlands). FACS-Lyse was obtained from Becton-Dickinson (Cowley, UK). Clinical grade unfractionated heparin (Leo Laboratories, UK), enoxaparin (Clexane; Rhône-Poulenc Rorer), lepirudin (Refludan, Pharmion) and bivalirudin (Angiox, Nycomed) were used in all studies.
Immunolabelling and flow cytometry
Platelet–monocyte aggregates were measured as previously described.2 Briefly, 60 µl aliquots of whole blood were incubated with anti-CD14, anti-CD42a and isotype-matched controls for 20 min at room temperature, before 500 µl of FACS-Lyse was added and 2500 cells were measured by flow cytometry (EPICS XL; Beckman Coulter, High Wycombe, UK). Monocytes were identified by their typical light scattering and CD14 expression. Platelet–monocyte aggregates were defined as monocytes positive for CD42a. To evaluate CD62P on platelets, blood was diluted 1:10 with phosphate-buffered saline and incubated with anti-CD42a, anti-CD62 and isotype-matched controls for 20 min at room temperature before further diluting the cells 1:30 with 1% paraformaldehyde. Platelets were identified by gating for CD42a and 5000 cells were measured. All results are expressed as percentage of positive cells. Data were analysed with EXPO 32 software (Beckman Coulter).
Statistical analysis
Continuous variables are reported as mean (SEM). Data were statistically analysed with Student’s t test or one-way analysis of variance where appropriate. All calculations were done with GraphPad Prism (V.3.02; Graph Pad Software, San Diego, California, USA). Significance was taken at 5%.
RESULTS
Effect of in vitro addition of anticoagulants on platelet–monocyte binding
Addition of unfractionated heparin (1 U/ml) in vitro was associated with a higher level of platelet–monocyte aggregates than in controls (20.1 (1.9)% v 16.2 (1.6)%, p < 0.001). However, platelet–monocyte aggregation was not affected by the addition of enoxaparin or lepirudin (16.9 (2.0)% and 17.0 (2.2)%, respectively, NS) (fig 1).
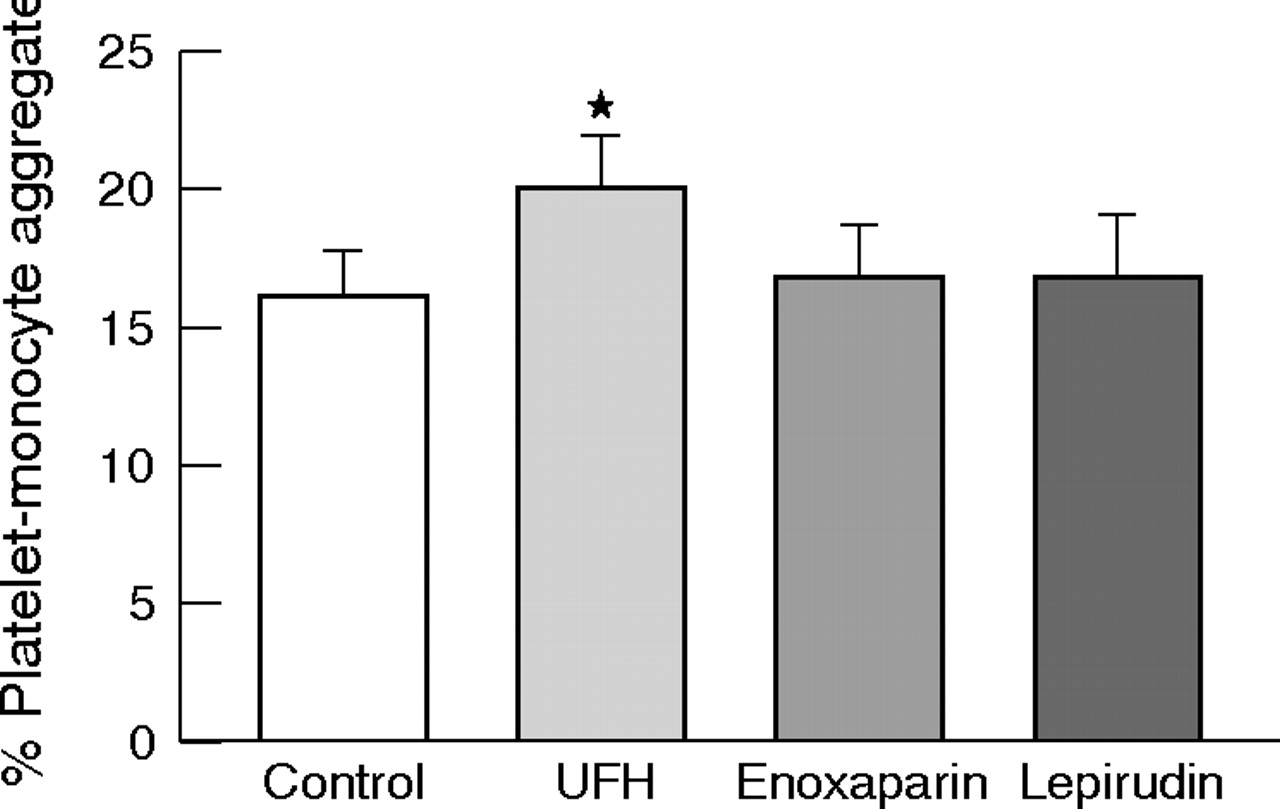
Effect of unfractionated heparin (UFH), enoxaparin and lepirudin on platelet–monocyte aggregates in vitro. Data are presented as mean (SEM). *p < 0.001 versus control, enoxaparin or lepirudin.
Mechanism of increased platelet–monocyte binding
Platelet surface expression of P-selectin was increased in whole blood treated with unfractionated heparin (6.9 (1.2)% v 3.2 (0.5)%, p < 0.01) (fig 2). Addition of P-selectin-blocking antibodies (CLB-thromb/6, 10 µg/ml) reduced platelet–monocyte aggregates by 71% (7.0 (1.2)% v 24.0 (2.6)%, p < 0.001). The addition of unfractionated heparin after P-selectin blockade was not associated with an increase in platelet–monocyte aggregates (6.4 (1.5)% v 7.0 (1.2)%, NS) (fig 3).
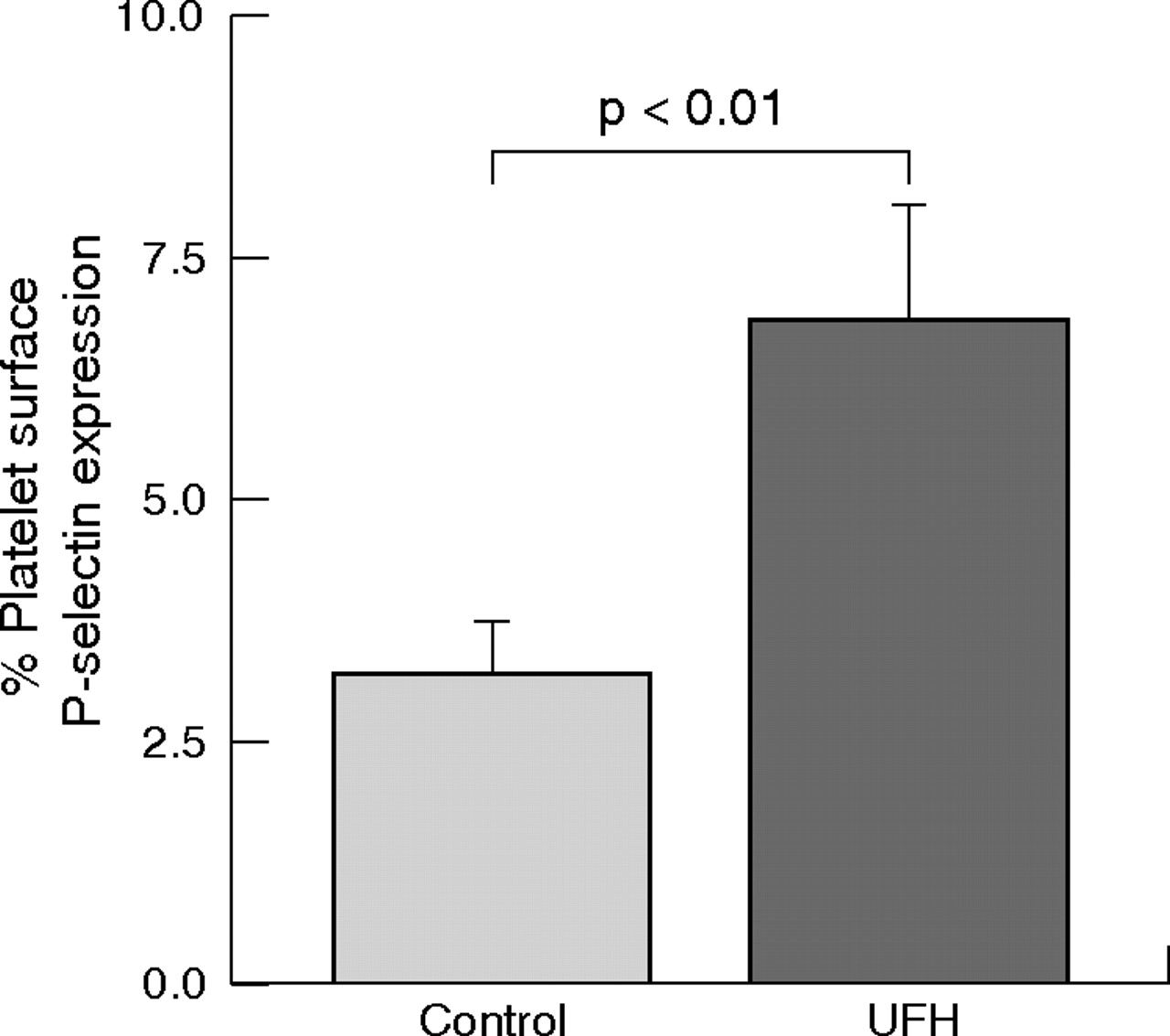
Effect of unfractionated heparin (UFH) on platelet surface P-selectin expression in vitro. Data are presented as mean (SEM).

Effect of unfractionated heparin (UFH) on platelet–monocyte aggregates with and without P-selectin (PS) blockade by anti-CD62P monoclonal antibody CLB-thromb/6 (final concentration 10 μg/ml). Data are presented as mean (SEM). *p < 0.05 versus controls; **p < 0.001 versus controls.
Effect of in vivo unfractionated heparin and direct thrombin inhibition on platelet–monocyte binding
To confirm our in vitro findings, we evaluated platelet–monocyte binding before and after giving unfractionated heparin and bivalirudin to patients scheduled for PCI and in a control group undergoing diagnostic coronary angiography with no anticoagulation. Platelet–monocyte aggregation did not differ at baseline between the three groups (mean 20.3 (1.7)%, p = 0.3). Consistent with our previous findings, in vivo administration of unfractionated heparin at routine clinical doses resulted in an increase in platelet–monocyte aggregates (absolute Δ 7.1 (2.7)%, p < 0.01). Platelet–monocyte binding did not change between the baseline and 5 min samples from patients receiving bivalirudin (absolute Δ −1.5 (2.4)%, p = 0.6) or the control group (absolute Δ 0.6 (0.7)%, p = 0.4) (fig 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Absolute change in platelet–monocyte aggregates after the administration of 100 U/kg of unfractionated heparin or 0.75 mg/kg of bivalirudin to patients scheduled for percutaneous coronary intervention and in a control population undergoing diagnostic coronary angiography with no anticoagulation. Data are presented as mean absolute change (SEM) from baseline. *p < 0.01 versus baseline.
DISCUSSION
We have shown that in vitro and in vivo unfractionated heparin is associated with platelet activation and an increase in platelet–monocyte aggregates through a P-selectin-dependent mechanism. This was not evident with direct thrombin inhibition or with fractionated heparin. These prothrombotic and proinflammatory effects of unfractionated heparin provide a potential explanation for the superior cardiovascular outcomes associated with fractionated heparins and direct thrombin inhibitors.
Platelet activation is pivotal in the pathogenesis of acute coronary syndromes and the complications of PCI and is associated with adverse outcomes.13,14 Some previous studies have suggested that use of unfractionated heparin may be associated with platelet activation.15–17 Consistent with these findings, our results showed that at therapeutic concentrations, unfractionated heparin increased platelet surface expression of P-selectin and platelet–monocyte aggregates, both of which are considered to be sensitive markers of platelet activation. In contrast, neither enoxaparin nor lepirudin increased platelet–monocyte aggregates in vitro, and the administration of bivalirudin in vivo did not affect platelet–monocyte aggregation.
The mechanism by which unfractionated heparin causes platelet activation in the absence of heparin-induced thrombocytopenia is uncertain. However, previous studies have shown that unfractionated heparin can bind to a wide range of receptors including the αIIbβ3 integrin (glycoprotein IIb/IIIa receptor), which in turn can result in outside-in signalling.18,19 This probably triggers a series of intracellular events leading to increased platelet activation.
Thrombosis and inflammation are closely related pathophysiological processes involving platelets, leucocytes and endothelial cells. Recently it has been appreciated that extensive cross talk occurs between these cells. Adhesion of activated platelets to monocytes induces nuclear translocation of nuclear factor-κB, the synthesis and expression of interleukin 1β, interleukin 8, monocyte chemoattractant protein 1, Mac-1, tissue factor and platelet-activating factor, and superoxide generation.6,7,8,9,10 Platelet–monocyte conjugation has been shown to promote monocyte adhesion to endothelium and recruitment to sites of arterial injury.11,12 Furthermore, Manka and colleagues20 reported that the absence of platelet P-selectin in apolipoprotein E-deficient mice significantly attenuated macrophage recruitment and neointimal formation after arterial injury. Thus, our findings suggest that unfractionated heparin, but not enoxaparin or direct thrombin inhibition, promotes platelet activation and platelet–monocyte interactions with proinflammatory and prothrombotic consequences. These findings may provide a mechanistic explanation for the trials showing that enoxaparin and direct thrombin inhibitors are superior to unfractionated heparin for the prevention of ischaemic events in patients presenting with acute coronary syndromes or those undergoing PCI.21–25
Heparin binds to a large number of proteins of differing structure and functionality.26 Previous in vitro studies have suggested that unfractionated heparin may modulate leucocyte function and exert an anti-inflammatory effect by binding to and blocking adhesion molecules including P-selectin, L-selectin and the integrin Mac-1 (CD11b/CD18).27–29 We have previously reported that platelet–monocyte conjugation is largely mediated by P-selectin expressed on the surface of activated platelets binding to the constitutively expressed monocyte receptor P-selectin glycoprotein ligand 1.2 Others have suggested that Mac-1 may also have an important role in platelet–leucocyte adhesion.30 Therefore, if unfractionated heparin effectively blocked P-selectin along with other adhesion molecules, one would expect a pronounced reduction in platelet–monocyte adhesion. In contrast, we found that treatment with unfractionated heparin increased platelet–monocyte binding both in vitro and in vivo. Furthermore, we found that P-selectin blockade reduced baseline platelet–monocyte aggregation by 71% and abolished any increase in platelet–monocyte binding after the addition of unfractionated heparin. Thus, our results suggest that unfractionated heparin does not inhibit P-selectin-based cell adhesion but rather promotes increased P-selectin-mediated adhesion between platelets and monocytes.
Conclusions
We have shown that platelet–monocyte aggregation is augmented in whole blood exposed to unfractionated heparin through a P-selectin-dependent mechanism. This was not evident in whole blood treated with enoxaparin or lepirudin, or in vivo after the administration of bivalirudin. Thus, unfractionated heparin not only activates platelets but also increases the adhesion of platelets to peripheral blood monocytes, with potential proinflammatory and prothrombotic consequences. Our findings provide a mechanistic explanation for the superiority of fractionated heparins and direct thrombin inhibitors over unfractionated heparin in patients with acute coronary syndromes or those undergoing PCI.
REFERENCES
Footnotes
-
Published Online First 18 May 2006
-
Drs Harding and Din were supported by grants from the British Heart Foundation (PG/2001/068; PG/03/009). This project was also supported by internal research funding from the Centre for Cardiovascular Sciences, University of Edinburgh.
-
Competing interests: Professor Fox has received grant support for the Global Registry of Acute Coronary Events (GRACE) from Sanofi-Aventis. All other authors have no conflict of interest to declare.