Article Text

Abstract
Background Fluticasone furoate/vilanterol (FF/VI) is a novel once-daily (OD) inhaled corticosteroid/long-acting β2 agonist combination in development for chronic obstructive pulmonary disease (COPD) and asthma.
Trial design A multicentre, randomised, double-blind, parallel-group, placebo-controlled study.
Methods Participants were patients with moderate-to-severe COPD treated with placebo or FF/VI 400/25 μg OD for 4 weeks. Study objectives were to assess the safety and efficacy of FF/VI 400/25 μg OD administered for 4 weeks via a novel dry powder inhaler. Co-primary end points were change from baseline in weighted mean (wm) heart rate 0–4 h postdose at day 28 and the incidence of adverse events (AEs). Secondary end points included change from baseline in trough forced expiratory volume in one second (FEV1) (23–24 h postdose; day 29) and wm FEV1 (0–4 h postdose; day 28). Patients were randomised to receive FF/VI 400/25 μg or placebo in a 2:1 ratio; all patients and investigators were blinded to active or placebo treatment.
Results 60 patients (mean age 64 years) were randomised (FF/VI: n=40; placebo: n=20), and all contributed data to the analysis. Mean screening post-bronchodilator FEV1 per cent predicted was comparable between groups (FF/VI: 58.5%; placebo: 60.1%). The wm heart rate 0–4 h postdose was similar between groups (difference: 0.6 beats per minute; 95% CI −3.9 to 5.1). More on-treatment AEs were reported in the FF/VI group (68%) compared with the placebo group (50%). The most common drug-related AEs in the FF/VI group were oral candidiasis (8%) and dysphonia (5%). There were no clinically relevant effects on laboratory values, including glucose and potassium, or on vital signs or ECGs/Holters. The FF/VI group had statistically greater improvements compared with placebo in trough FEV1 (mean difference 183 ml) and 0–4 h postdose wm FEV1 (mean difference 236 ml).
Conclusion FF/VI has a good safety and tolerability profile and improves lung function compared with placebo in patients with COPD.
Trial registration number clinical trials.gov—NCT00731822.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Article summary
Article focus
Is the once-daily inhaled corticosteroid/long-acting β2 agonist (ICS/LABA) combination FF/VI efficacious with a favourable safety and tolerability profile in COPD?
Key messages
In patients with moderate-to-severe COPD, FF/VI 400/25 μg once daily improved lung function. AEs frequently experienced with other ICS/LABA combinations were generally reported at similar frequencies in the placebo and active treatment arms.
Strengths and limitations of this study
This paper is the first to present clinical data on inhaled FF/VI combination therapy in patients with chronic obstructive lung disease. Given the 4-week duration of this study, there was no end point or surrogate marker to specifically address the relative clinical effects of FF in COPD (such as exacerbations), whereas the observed lung function effects are predominantly induced by the LABA component of the combination.
Introduction
Chronic obstructive pulmonary disease (COPD) is a significant cause of morbidity and mortality that contributes substantially to healthcare costs and morbidity worldwide.1 2 Unlike other chronic diseases, it is increasing in prevalence and is projected to be the fourth most common cause of death worldwide by 2030.3 Consequently, an unmet need continues to exist for therapies directed at reducing the morbidity and mortality of COPD.
Anti-inflammatory therapies administered in combination with bronchodilators according to disease severity are a key approach by which COPD can be managed in the long term,4 as they target both the inflammation and the bronchoconstriction that contribute to the pathophysiology of the disease.5–7 Long-term studies indicate that combination therapies consisting of a bronchodilatory long-acting β2 agonist (LABA) plus an anti-inflammatory inhaled corticosteroid (ICS) in one inhaler have the potential to modify disease progression through positive effects on lung function, symptoms and exacerbations.8–12
Current ICS/LABA combinations are dosed twice daily; however, once-daily treatment has the potential to simplify treatment in chronic disease such as COPD by reducing dosing frequency.13 Vilanterol (VI) and fluticasone furoate (FF) are, respectively, a novel inhaled LABA and ICS in development for once-daily combination therapy for COPD and asthma. VI is an antedrug analogue of salmeterol with a higher intrinsic activity at the β2 receptor than salmeterol.14 In vitro, VI exhibits >1000 fold selectivity for β2 receptors relative to β1 or β3 receptors,15 while data from human lung tissue indicate a faster onset and longer duration of action (22 h) than salmeterol.16 FF is chemically distinct from fluticasone propionate (FP) in that the 17α-ester of the fluticasone moiety comprises a furoate, as opposed to propionate group; this group is not cleaved from the molecule during metabolism.17 In vitro, studies of FF suggest a pharmacological profile that differs from FP and other ICS; FF exhibits greater potency in cell culture models of inflammation compared with FP and budesonide, shows greater potency compared with FP in peripheral blood mono-nuclear cells from patients with mild asthma or moderate/severe COPD and is further differentiated from FP in that cell culture assays of glucocorticoid-dependent gene expression and glucocorticoid receptor nuclear translocation indicate activity at >24 h, which is not observed with FP.18 Clinically, preliminary results in patients with COPD have demonstrated that VI is well tolerated with a good safety profile.19 20
This is the first study to report on the clinical effects of the combination of FF/VI in patients with obstructive lung disease. The aim was to assess the short-term safety, tolerability and efficacy of FF/VI in patients with COPD, with a primary focus on safety; efficacy was a secondary end point. Preliminary results from this study have been presented in abstract form.21
Methods
Patients
The study was conducted between August 2008 and February 2009 at nine centres in two countries (Norway and Sweden). To be eligible, patients were required to be aged between 40 and 80 years, exhibiting a clinical history of COPD, have a smoking history of ≥10 pack-years, a post-bronchodilator forced expiratory volume in one second (FEV1)/forced vital capacity ratio of ≤0.70 and a post-bronchodilator FEV1 ≥40% and ≤80% predicted (GOLD stage II/III).
Patients were allowed ipratropium bromide for use as rescue medication during the screening period and throughout the study treatment period. Other permitted medications during the screening and the treatment periods included: intranasal sodium cromoglycate or nedocromil sodium, mucolytics, antibiotics (for short-term treatment of acute non-respiratory tract infections), antihistamines, cough suppressants (for short-term treatment ≤7 days), intranasal corticosteroids (patients were to be on a stable, daily dose for at least 4 weeks prior to visit 1 and throughout the study); topical or ophthalmic corticosteroids.
Medications prohibited prior to and during the study included: depot corticosteroids (12 weeks prior to screening); systemic, oral or parenteral corticosteroids (6 weeks prior to screening); p-glycoprotein inhibitors (6 weeks prior to screening); cytochrome P450 3A4 strong inhibitors (6 weeks prior to screening); ICS alone or in combination with a LABA (4 weeks prior to screening); tiotropium (1 week prior to screening); theophyllines, LABA (oral and inhaled) and oral leukotriene inhibitors (48 h prior to screening); inhaled sodium cromoglycate or nedocromil sodium (24 h prior to screening); ipratropium/albuterol combination products and short-acting β2 agonists (6 h prior to screening); diuretics; monoamine oxidase inhibitors; β receptor antagonists; anticonvulsants and phenothiazines; tricyclic antidepressants.
Patients were excluded if they exhibited: any respiratory disorder other than COPD; poorly controlled COPD defined as having acute worsening (patient-managed corticosteroid or antibiotic treatment or physician prescription) within 6 weeks of the study or a hospitalisation for COPD within 12 weeks; a lower respiratory tract infection requiring treatment with antibiotics within 6 weeks of the study; requirement for daily long-term oxygen therapy (≥12 h/day). No exclusion criteria were imposed based on reversibility to 400 μg albuterol administered via metered-dose inhaler. According to the study protocol, reasons for withdrawal from the study as recorded in the electronic Case Report Form included: adverse event (AE), lack of efficacy, protocol deviation, lost to follow-up, investigator discretion, withdrawal of consent, patient met any of the liver chemistry stopping criteria, patient provided a positive serum pregnancy test or was repeatedly non-compliant (<80%) with study medication. Patients could also be withdrawn from the study if any of the following criteria were met: COPD exacerbation (defined as worsening of COPD symptoms requiring the use of any treatment other than study medication or rescue ipratropium); clinically important changes in laboratory parameters; pneumonia (confirmed by chest x-ray within 48 h of diagnosis); Electrocardiogram (ECG) abnormalities including maximum resting heart rate of ≥140 beats per minute (bpm) or an increase on heart rate from baseline of ≥40 bpm (two measure ≥5 min apart), prolongation in absolute QTc(B(Bazett's formula)) to >500 ms (>600 ms uncorrected QTc; confirmed by three readings ≥5 min apart on ≥3 ECGs), ventricular rate <37 bpm (confirmed by three readings ≥5 min apart on ≥3 ECGs), PR interval >240 ms (confirmed by three readings ≥5 min apart on ≥3 ECGs), pathological Q waves, non-specific intraventricular conduction delay, ST-T wave abnormalities (excluding non-specific ST-T wave abnormalities), right or left complete bundle branch block.
All patients gave written informed consent, and the protocol was approved by the appropriate institutional review boards and conducted in accordance with good clinical practice guidelines and the 1996 version of the Declaration of Helsinki.
Study design
This was a 4-week, multicentre, randomised, placebo-controlled, parallel-group study (GSK study number HZC111348; clinical trials.gov NCT00731822). Patients entered a ≤7-day screening period to collect baseline safety data and to determine eligibility; eligible patients were then randomised 2:1 to inhaled FF/VI combination (400/25 μg) or placebo administered once daily in the morning by a novel dry powder inhaler (NDPI; Glaxo-SmithKline, London, UK). This was a double-blind study. Neither the subject nor the investigator knew which study medication the subject was receiving; the NDPI devices containing active medication or placebo appeared identical to the patient, their caregivers and the investigator, and the placebo dry powder formulation was indistinguishable from the FF/VI inhalation powder formulation. The central randomisation schedule was generated by the sponsor using a validated computerised system (RandAll). Patients were randomised using the Registration and Medication Ordering System (RAMOS), an automated interactive telephone-based system that was used by the investigator or designee to register the patient, randomise the patient and receive medication assignment information. Patients were eligible to be randomised provided none of the following occurred between visit 1 (screening) and visit 2 (randomisation): COPD exacerbation or lower respiratory tract infection; abnormal clinically significant findings including liver chemistry, biochemical, haematology or urinalysis screening tests; evidence of active, clinically significant abnormalities in 24-h Holter ECG at screening or predose 12-lead ECG at visit 2; evidence of non-compliance with prohibited medications and keeping clinic visit appointments. Post-randomisation, patients were reviewed at days 2, 7 (telephone contact), 14, 15, 28 and 29. All visits occurred in the morning with visits on days 2, 15 and 29 needing to occur within 22 h following study medication administration to obtain the 23–24-h postdose trough FEV1 measurement. Patients withheld study medication on the morning of clinic visits and rescue medication for at least 6 h prior to clinic visits.
A patient who had been assigned a patient identifier and had completed at least one study procedure (in addition to signing a consent form) but had not met inclusion or randomisation criteria to enter the treatment period or who did meet the exclusion criteria was classified as a screening failure.
The 400 μg dose of once-daily FF was chosen as this dose had been previously assessed for up to 8 weeks in studies of FF monotherapy in patients with asthma and had no clinically significant effect on the hypothalamic–pituitary–adrenal axis.22–25 VI at 25 μg has demonstrated sustained bronchodilation in preliminary studies in patients with COPD.19 26
Outcome measurements
The co-primary safety end points were the change from baseline in supine (lying down with head upward) weighted mean heart rate 0–4 h postdose at the end of the 28-day treatment period and the incidence of AEs throughout. Heart rate was assessed using standardised ambulatory monitors (Omron HEM-711DLX device (Omron Healthcare Inc. Kyoto, Japan) supplied by Biomedical Systems (Brussels, Belgium, and Maryland Heights, Missouri, USA)) throughout the study to ensure accuracy and consistency of data recorded. Vital signs were collected predose and at 15, 45, 90 min and 2 and 4 h postdose on days 1, 14 and 28 and predose for all other visits. AEs (coded using the Medical Dictionary for Regulatory Authorities) were assessed during each clinic visit. Each patient was provided with a ‘medical problems/medications taken diary’ in which they were instructed to record any medical problems that they experienced and any medications used to treat the medical problem(s). The investigator or designee reviewed the diary at each clinic visit, and all confirmed AEs/serious AEs were recorded in the electronic Case Report Form.
Other safety end points included weighted mean and maximum systolic blood pressure and mean arterial blood pressure and weighted mean and minimum diastolic blood pressure over 0–4 h on days 1, 14 and 28. Weighted mean glucose and potassium over 0–4 h on days 1, 14 and 28 and maximum glucose and minimum potassium were other safety end points. Weighted mean and maximum QTc(F) (QT interval corrected by Fridericia's method) and QTc(B) (QT interval corrected by Bazett's method) derived from 12-lead ECGs recorded predose and 30 min and 1, 2 and 4 h postdose on days 1, 14 and 28, changes in haematologic and clinical chemistry parameters on days 14 and 28 and 24-h, 3-lead Holter ECG measurements at screening and day 28 were additional safety end points. Standardised ECG (PCECG 1200M ECG machine) and 3-lead Holter devices (DATRIX DR512 VX3 (Biomedical Systems, Brussels, Belgium, and Maryland Heights, Missouri, USA)) were used throughout the study, with central reader over-reads performed (representative from Biomedical Systems) to ensure consistency and accuracy of data recorded. Collection supplies for blood and blood analyses (for routine assessment of chemistry and haematology analytes and the pharmacodynamic assessments of plasma glucose and serum potassium levels) and urine collection supplies and sample analyses were provided by a central laboratory (Quest Diagnostics Clinical Trials (Heston, Middlesex, UK)). The incidence of COPD exacerbations was also recorded throughout the study. Each patient was provided with an electronic peak flow meter (Micro Medical MicroDiary electronic peak flow meter provided by Biomedical Systems, Brussels, Belgium), which the patient used to measure their peak expiratory flow three times, twice daily, in the morning and evening from screening to the end of 4-week treatment period.
Secondary efficacy end points were assessed by clinic spirometry. All sites used standardised spirometric equipment (Vitalograph Centralized Spirometry System (Biomedical Systems, Brussels, Belgium, and Maryland Heights, Missouri, USA)), and data were over-read by a trained over-reader (representative from Biomedical Systems) to ensure consistency and accuracy of collected data. Secondary efficacy end points were change from baseline in trough clinic visit FEV1 (23–24 h postdose) on days 2, 15 and 29 (to assess the 24 h effect of VI and to evaluate the contribution of FF) and weighted mean FEV1 (0–4 h postdose) on days 1 and 28 (to evaluate the contribution of VI). Trough FEV1 was defined as the mean of the FEV1 values obtained 23 and 24 h after dosing on days 1, 14 and 28. Other end points included time to an increase of ≥100 ml above baseline in FEV1 on day 1 (0–4 h), peak FEV1 on days 1 and 28 (0–4 h) and serial FEV1 and forced vital capacity on days 1 and 28 (to evaluate the contribution of VI).
Statistical analysis
The sample size of 60 evaluable patients was based on the co-primary end point of change from baseline in weighted mean heart rate 0–4 h postdose at the end of the 28-day treatment period. The expected variability used for the sample size calculations in this study was based on data from a previous COPD study (B2C108562) with the compound GW642444H (data not shown), which demonstrated between-patient estimates of standard deviation (SD) of up to 10 bpm for change from baseline weighted mean heart rate. The residual SD from the corresponding statistical analyses was in the range of 7.2–8.0 bpm. In the current study, weighted mean heart rate was calculated from heart rate recorded at clinic visits and so a more conservative estimate of SD of 10 bpm was used for the sample size calculations. FF/VI was considered non-inferior to placebo for heart rate if the upper limit of the 95% CI for the estimated treatment difference for weighted mean heart rate was less than +10 bpm. The true difference between placebo and FF/VI was assumed to be 0 bpm. The number of completers in the B2C108562 study was 17 in the placebo group and 34 in the GW642444H group; therefore, if a similar number of patients completed the current study, there would be a 90% power to show non-inferiority to placebo. To allow for a possible withdrawal rate of 15%, 20 patients were randomised to placebo and 40 to FF/VI.
All efficacy and safety analyses used the intent-to-treat (ITT) population, defined as all randomised patients who received at least one dose of study medication. The primary analysis was performed for the ITT population using a Repeated Measures Mixed Model for weighted mean heart rate 0–4 h postdose on days 1, 14 and 28 (visits 2, 5 and 7). A one-sided 2.5% risk associated with incorrectly rejecting the null hypothesis (significance level) was considered acceptable for this study. As this was a single comparison, and there was no planned formal statistical analysis for the AE data, no adjustment was made for multiplicity.
Pair-wise comparisons between FF/VI and placebo were performed for all secondary efficacy end points and other key end points using Repeated Measures Mixed Models for the ITT population.
Results
Of 89 patients screened, 60 were randomised and comprised the ITT population. The reasons for screening failure were failure to meet inclusion/exclusion criteria (n=16), failure to meet randomisation criteria (n=8) and withdrawal of consent (n=5). Details of patient disposition and reasons for discontinuation are shown in figure 1, and patient baseline characteristics are shown in table 1.
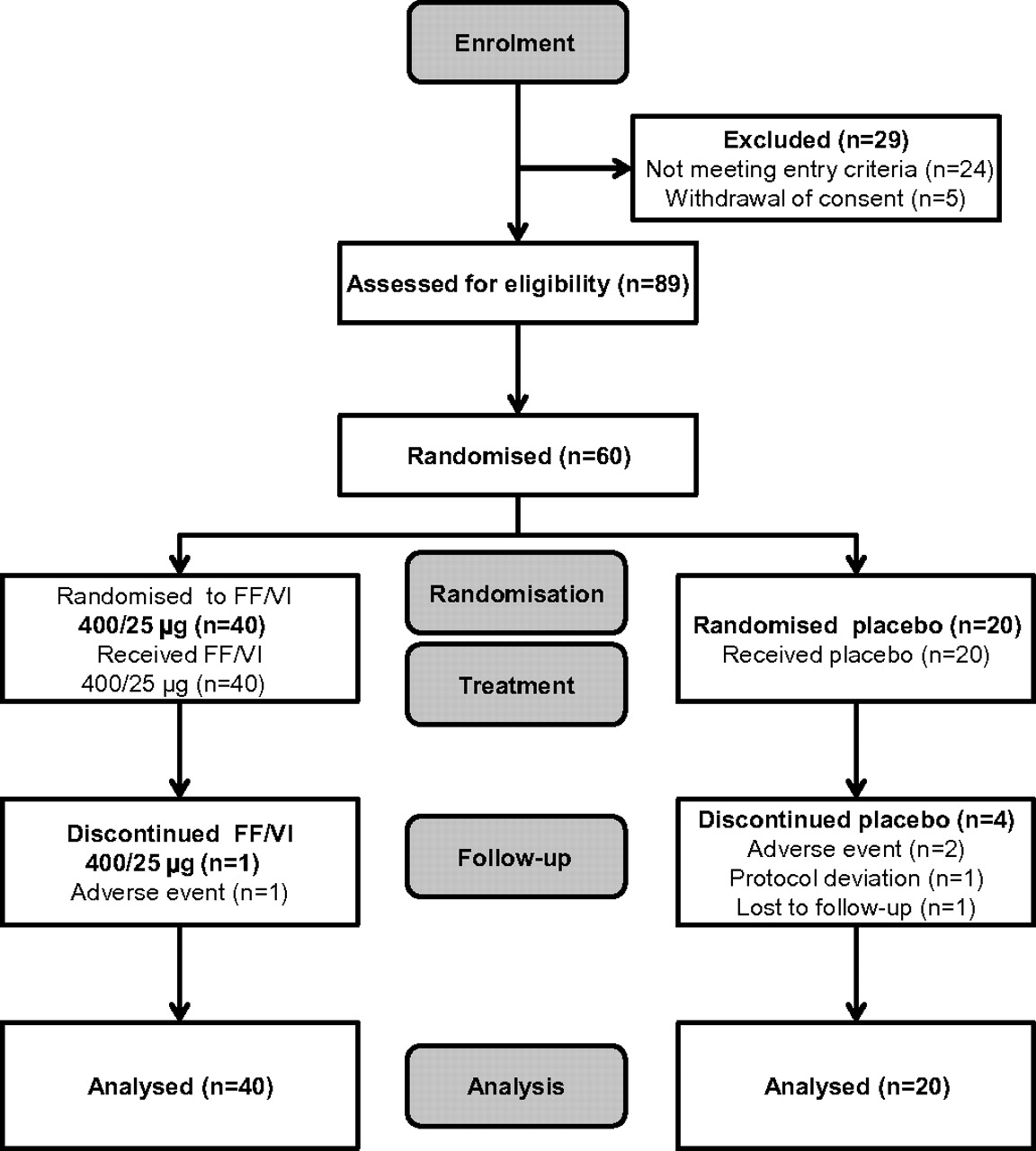
Patient disposition and reasons for discontinuation for the fluticasone furoate (FF)/vilanterol (VI) (400/25 μg) and placebo treatment groups.
Patient demographics (intent-to-treat population)
The history of COPD was similar across the treatment groups, with the majority of the ITT population (87%) having a COPD diagnosis of <10 years. Smoking history was similar across the treatment groups, with 37% of the overall ITT population being current smokers (40% in the placebo group and 35% in the FF/VI group). During the run-in period, 11 patients (55%) in the placebo group and 15 patients (38%) in the FF/VI group used COPD medication. Short-acting bronchodilators (ipratropium bromide/salbutamol) were the most frequently used medications; ICS/LABA combination therapy was used by one patient (5%) in the placebo group and by two patients (6%) in the FF/VI group. Eight patients (40%) in the placebo group and 14 patients (35%) in the FF/VI group were taking concomitant COPD medication during the treatment period. The most common medication was ipratropium bromide, taken by six patients (30%) in the placebo group and by 11 patients (28%) in the FF/VI group. The mean treatment exposure was similar across the two treatment groups, with 80% of patients in each group exposed to study treatment for between 28 and 30 days.
Safety
There was no statistically significant difference in the adjusted mean change from baseline in weighted mean heart rate 0–4 h postdose between the FF/VI and placebo groups (+0.7, +0.8 and +0.6 bpm on days 1, 14 and 28, respectively). FF/VI was non-inferior to placebo as the upper CI was below the predefined non-inferiority limit of +10 bpm. Figure 2 shows adjusted mean change from baseline for postdose 0–4 h weighted mean heart rate for FF/VI and placebo at days 1, 14 and 28.

Adjusted mean change from baseline in weighted mean (wm) heart rate 0–4 h postdose on days 1, 14 and 28 (intent-to-treat population). FF, fluticasone furoate; VI, vilanterol.
There were no statistically significant differences between the FF/VI and placebo groups on maximum heart rate, weighted mean systolic blood pressure or maximum systolic blood pressure as the 95% CI for all differences contained zero (table 2).
Adjusted mean differences from PBO (95% CI), safety analysis (intent-to-treat population)
For the weighted mean diastolic blood pressure and minimum diastolic blood pressure, there was no consistent evidence of a treatment effect. On both days 1 and 28, the 95% CI values for the mean change from baseline in these two parameters included zero, indicating no difference from placebo. On day 14, the 95% CI values did not include zero, suggesting a difference from placebo (table 2). The changes from baseline in the weighted mean for the mean arterial blood pressure were minimal and similar between the placebo and FF/VI groups on days 1, 14 and 28.
The number and proportion of patients who reported an AE while receiving study treatment was 27 (68%) in the FF/VI group and 10 (50%) in the placebo group. Of these patients, nine (23%) in the FF/VI group and two (10%) in the placebo group experienced AEs considered to be possibly related to the study drug. Nasopharyngitis was the most common on-treatment AE reported in both the placebo and the FF/VI groups (three (15%) patients in placebo group and seven (18%) patients in FF/VI group). The most frequent on-treatment AEs are shown in table 3. Oral candidiasis and dysphonia were the most common drug-related AEs reported in the FF/VI group (three patients (8%) and two patients (5%), respectively). No deaths were reported. One non-fatal serious AE (ulcerative colitis) was reported in the FF/VI group by a patient with a medical history of ulcerative colitis. The study investigator considered that there was no reasonable possibility that the ulcerative colitis may have been caused by the investigational drug. Withdrawals due to AEs were low in both the FF/VI group (n=1 patient: diverticulum) and the placebo group (n=2 patients: an abnormal ECG (considered to be treatment related) and urinary tract infection). No COPD exacerbations were experienced in either treatment group.
On-treatment adverse events experienced by ≥5% of patients in any treatment group (intent-to-treat population)
A small statistically significant increase (0.32 mmol/l; 95% CI 0.10 to 0.53) with FF/VI versus placebo was observed on day 28 (but not on days 1 and 14) for change from baseline in weighted mean plasma glucose FF/VI (figure 3A). No statistically significant difference was seen between the treatment groups for the change from baseline in weighted mean potassium (figure 3B). Mean QTc(F(Fridericia's correction)) intervals predose on day 1 were similar between the placebo and FF/VI groups (398 and 392 ms, respectively). The mean changes from baseline in QTc(F) at days 1, 14 and 28 were small and ranged from −2.9 to 3.2 ms in the placebo group and from −5.9 to 3.6 ms in the FF/VI group (figure 4). For weighted and maximum 0–4 h QTc(F), no significant treatment difference in adjusted mean change was observed at any time point between treatments. All patients in both treatment groups had maximum postdose QTc(F) intervals of ≤450 ms.

Adjusted mean change from baseline (0–4 h) weighted mean (wm) glucose (A) and potassium (B) (intent-to-treat population). FF, fluticasone furoate; VI, vilanterol.

Adjusted mean change from baseline in weighted mean (wm) QTc(F) (0–4 h) (intent-to-treat population). FF, fluticasone furoate; VI, vilanterol.
There were also no apparent trends for a treatment effect on premature ventricular beats, supraventricular beats or ventricular runs, as measured by a 3-lead Holter device. No clinically relevant treatment effects were observed on laboratory parameters. Results for AM and PM peak expiratory flow during the screening and study periods (days 1–28) are shown in table 4.
Summary results for AM and PM PEF (screening period and days 1–28)
Efficacy
The 95% CI values for the adjusted mean difference between placebo and FF/VI in change from baseline in trough FEV1 on days 2, 15 and 29 all excluded zero (table 5), indicating a statistically significant improvement in trough FEV1 with FF/VI compared with placebo. Adjusted mean change from baseline trough FEV1 values for FF/VI and placebo on days 2, 15 and 29 is shown in figure 5A.
Changes in lung function from baseline in trough and wm (0–4 h) FEV1 (l)

Adjusted mean change from baseline in (A) trough forced expiratory volume in one second (FEV1) at days 2, 15 and 29, and (B) weighted mean (wm) (0–4 h) FEV1 on days 1 and 28 (intent-to-treat population). FF, fluticasone furoate; VI, vilanterol.
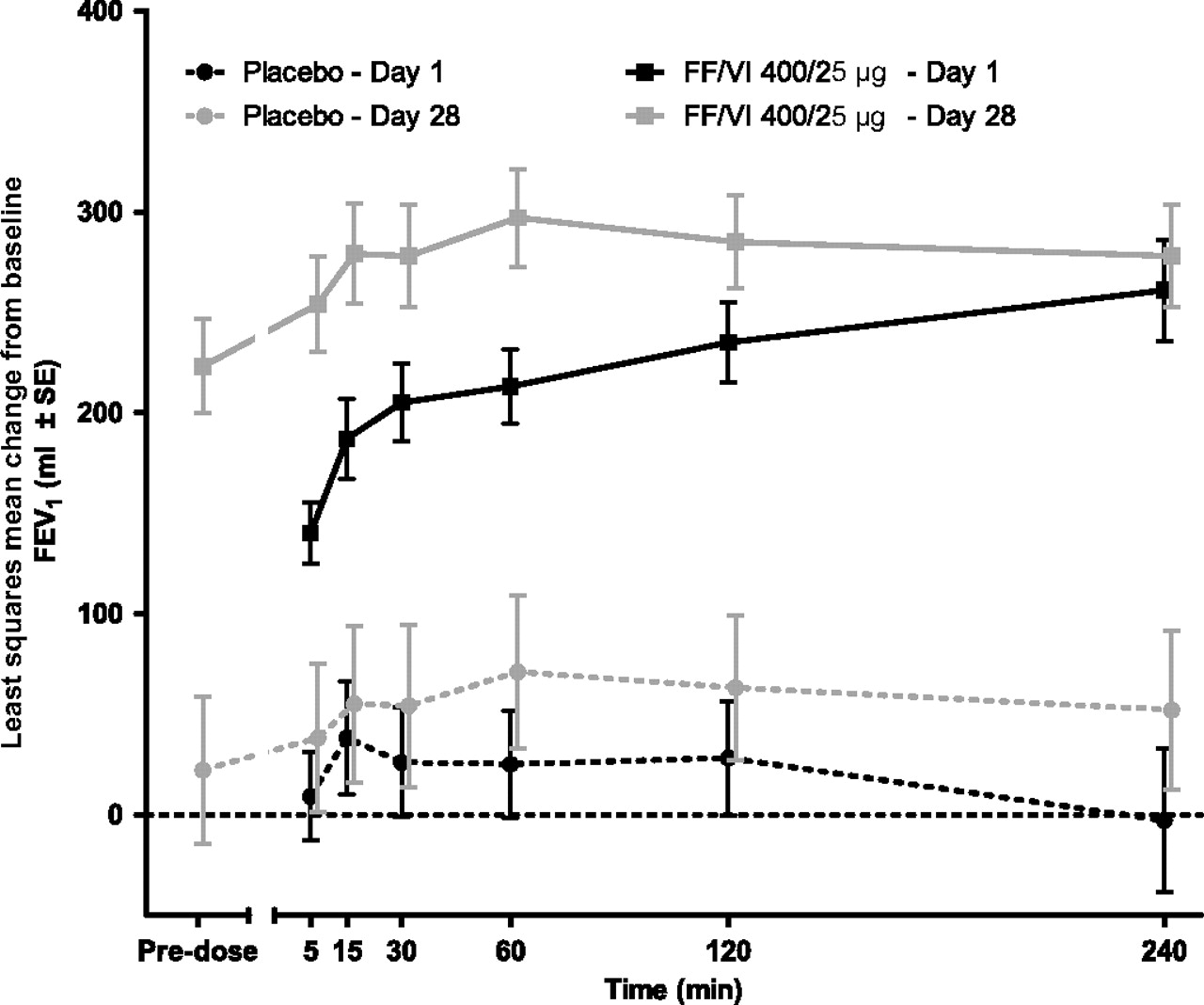
Similarly, adjusted mean treatment differences in change from baseline in weighted mean FEV1 (0–4 h) on days 1 and 28 showed statistically greater improvements of FEV1 in the FF/VI group relative to placebo (table 5). Adjusted mean change from baseline weighted mean (0–4 h) FEV1 values for FF/VI and placebo on days 1 and 28 is shown in figure 5B. The adjusted mean treatment differences in change from baseline in 0–4 h serial FEV1 on days 1 and 28 were higher for the FF/VI group compared with the placebo group at all time points; the CIs did not include zero, indicating a statistically significant difference across the time points (table 6 and figure 6). The majority (65%) of patients in the FF/VI group achieved a ≥100 ml increase from baseline FEV1 at the 5 min postdose time point on day 1, the first postdose time point at which FEV1 was measured, compared with 10% in the placebo group. By the 4 h postdose time point, 85% of patients in the FF/VI group and 50% of patients in the placebo group had achieved a ≥100 ml increase from baseline (table 7). The mean changes from baseline in peak FEV1 (0–4 h postdose) were higher on both days 1 and 28 for the FF/VI group (315 and 348 ml, respectively) compared with the placebo group (98 and 119 ml, respectively).
Serial changes in lung function 0–4 h postdose forced expiratory volume in one second (l) on days 1 and 28

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Least squares mean change from baseline in serial forced expiratory volume in one second (FEV1) on days 1 and 28 (intent-to-treat population). FF, fluticasone furoate; VI, vilanterol.
Time to patients achieving their first instance of ≥100 ml improvement in forced expiratory volume in one second 0–4 h postdose day 1 (intent-to-treat population)
Discussion
This paper is the first to present clinical data on inhaled FF/VI combination therapy in patients with chronic obstructive lung disease. FF/VI, dosed once daily, did not affect heart rate, had an on-treatment AE profile currently known to be associated with ICS therapy and showed clinical efficacy compared with placebo.
The results of the safety evaluation of this study suggest that FF/VI has a clinically acceptable safety profile at the given dose (400/25 μg once daily). Previous studies of other twice-daily LABAs in both patients and healthy volunteers have shown that they can be associated with β2-mediated systemic adverse effects such as headache, palpitations with changes in heart rate, prolongation of the QTc interval and increased glucose and/or decreased potassium blood levels.27–29 Consequently, these parameters were investigated in this study. The lack of difference between the FF/VI and placebo groups in the adjusted mean change from baseline in weighted mean heart rate 0–4 h postdose provides strong evidence of a lack of clinical effect of FF/VI on heart rate. There was also no evidence of clinically relevant treatment effects in the ECG assessments, including dysrhythmias. The absence of detrimental cardiovascular incidents in this study suggests that the risk of adverse cardiovascular effects with FF/VI is low. Longer term studies in substantially larger cohorts of patients with COPD would be required to determine whether any subgroup of patients with COPD may be at higher risk of developing cardiac side effects. Nevertheless, given that cardiovascular diseases are more prevalent in patients with COPD30 and there is an ongoing discussion related to the cardiac safety of many COPD therapies,31–33 it is notable that no cardiac safety signal was detected in this study. No clinically relevant treatment effects on haematology, clinical chemistry or urinalysis were observed. A statistically significant difference between FF/VI and placebo was observed at day 28 (but not on days 1 and 14) for weighted mean plasma glucose; this difference was driven by a decrease in the mean change from baseline in plasma glucose in the placebo group (see figure 3A) rather than an increase in plasma glucose in the FF/VI group.
The proportion of patients experiencing one or more on-treatment AEs was higher in the FF/VI group (68%) compared with the placebo group (50%), and these events were primarily accounted for by AEs commonly reported with ICS treatment (nasopharyngitis and oral candidiasis). Dysphonia, an expected AE with ICS treatment, was observed at the incidence of 5% in the FF/VI group and was not observed in the placebo group. No COPD exacerbations were reported during the study.
The results of the secondary efficacy analysis showed a significant improvement in FEV1 relative to placebo with FF/VI treatment. A treatment difference of 183 ml in trough FEV1 measured 23–24 h postdose was observed on day 29, showing a 24-h duration of effect for this combination. This improvement was greater than the 100–140 ml outcome recently suggested to be of a minimal important difference in clinical trials of COPD.34 Serial measurements of FEV1 on day 1 showed a rapid onset of action for FF/VI compared with placebo. Clinically relevant bronchodilation (≥100 ml) was observed at the 5-min postdose time point following the first dose (the earliest postdose assessment time point in this study) in most patients receiving FF/VI.
A sustained 24-h effect is advantageous in the treatment of patients with COPD, providing it is not accompanied by a loss in efficacy during chronic dosing, as could be seen if tolerance to the bronchodilating effect was induced. Mean changes from baseline in maximal weighted mean FEV1 observed 0–4 h postdose were higher on both days 1 and 28 for FF/VI compared with placebo, which argues for no loss in efficacy over the 4-week treatment period and hence an absence of tolerance. It is unlikely that longer term studies with regular treatment would detect any induction of tolerance, as any measurable β2 agonist-induced tolerance is typically observed within a few days of first dosing.35
There are some limitations of this phase IIa study. Most importantly, there was no end point or surrogate marker to specifically address the relative clinical effects of FF in COPD (such as exacerbations), whereas the observed lung function effects are predominantly induced by the LABA component of the combination. Related to this, nor was VI assessed as a monotherapy. As exacerbations are a major cause of morbidity,36 a study with a much larger sample size and performed over a longer period of time is required to determine the effects of FF/VI on exacerbation frequency. This raises the second main limitation of this study, which is the 28-day treatment course. Although sufficient to assess the co-primary safety end points and typical for a phase IIa study, the 28 days of treatment in this study is not a sufficient period of time from which firm conclusions, as to the long-term efficacy of FF/VI, can be made. Another issue associated with the ICS component of the FF/VI combination investigated is the potential impact of ICS treatment on the hypothalamic–pituitary–adrenal axis. Morning urinary cortisol excretion may be decreased during ICS/LABA therapy, and urinary cortisol excretion was not measured in this study. Although only one dose of FF/VI was investigated, a previous dose-ranging study investigating the effect of 3, 6.25, 12.5, 25 and 50 μg of VI over 4 weeks has demonstrated a good safety and efficacy profile in patients with COPD.20 26
In conclusion, this study showed that FF/VI 400/25 μg administered once daily by an NDPI was efficacious with a favourable safety and tolerability profile. Combination therapy in COPD is established to be an effective treatment option in moderate-to-severe cases of disease, and these results indicate that a once-daily regimen of this combination is a possible means of addressing the unmet needs in COPD.
Acknowledgments
All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. We thank all patients who took part in the study and the following HZC111348 study investigators: Ragnar Dahle, M.D. (Fredrikstad, Norway), Sigbjørn Elle, M.D. (Elverum, Norway), Per Arne Lier, M.D. (Sandvika. Norway), Bo Lundbäck, Ph.D. (Lulea, Sweden), Michael Runold, M.D., Ph.D. (Stockholm, Sweden) and their staff. We also thank Jie Wang, M.D., Ph.D., GlaxoSmithKline Inc., for her contributions during protocol development and during the study.
Editorial support in the form of development of draft outline, development of manuscript first draft, editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments, copyediting, fact checking, referencing and graphic services was provided by Geoff Weller at Gardiner-Caldwell Communications and was funded by GlaxoSmithKline. The article-processing charge was paid by GlaxoSmithKline.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data Supplement - Manuscript file of format pdf
Footnotes
To cite: Lötvall J, Bakke PS, Bjermer L, et al. Efficacy and safety of 4 weeks' treatment with combined fluticasone furoate/vilanterol in a single inhaler given once daily in COPD: a placebo-controlled randomised trial. BMJ Open 2012;2:e000370. doi:10.1136/bmjopen-2011-000370
Funding This study was funded by GlaxoSmithKline, study number HZC111348.
Competing interests JL has served as a consultant to and received lecture fees from AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme, Novartis and UCB Pharma; has been partly covered by some of these companies to attend previous scientific meetings including the ERS (European Respiratory Society annual congress) and the AAAAI (American Academy of Allergy Asthma and Immunology annual meeting); and has participated in clinical research studies sponsored by AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme, and Novartis. PSB has received lecture fees from AstraZeneca, GlaxoSmithKline and NycoMed; has participated in clinical research studies sponsored by GlaxoSmithKline, Pfizer and Boehringer Ingelheim; and is currently member of the Steering Committee and the Scientific Committee of the ECLIPSE study, which is sponsored by GlaxoSmithKline. LB has served as a consultant to and received lecture fees from AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme, Novartis and UCB Pharma; has been partly covered by some of these companies to attend previous scientific meetings including the ERS and the AAAAI; and has participated in clinical research studies sponsored by AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme, and Novartis. SS has been sponsored by AstraZeneca, GlaxoSmithKline, Boehringer Ingelheim/Pfizer to attend meetings and congresses including ATS (American Thoracic Society annual meeting) and ERS; has participated in clinical research studies sponsored by AstraZeneca, GlaxoSmithKline and Boehringer Ingelheim; and has received lecture fees from GlaxoSmithKline and Boehringer Ingelheim. CS-W, CC, LS, BH are employees of and hold stock in GlaxoSmithKline.
Ethics approval Ethics approval was provided by institutional review board.
Contributors All authors developed the design and concept of the study, had full access to and interpreted the data, and wrote the manuscript. LS, CS-W, BH and CC approved the statistical plan. CS-W coordinated data gathering. LS led the statistical analysis. All authors vouch for the accuracy and completeness of the data and the data analysis. The independent steering committee (JL, PSB, LB and SS) together with the authors employed by the sponsor (CS-W, CC, LS and BH) had full access to the data and were responsible for the decision to publish the paper. Employees of the sponsor performed the statistical analysis, led by LS. The sponsor did not place any restriction on authors about the statements made in the final paper.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Additional data supporting the reported results and conclusions of this study have been made available at http://datadryrad.org/ with the following doi:10.5061/dryad.7p1r30q5