


Abstract
The transient receptor potential (TRP) vanilloid subtype 4 (V4) is a nonselective cation channel that exhibits polymodal activation and is expressed in the endothelium, where it contributes to intracellular Ca2+ homeostasis and regulation of cell volume. The purpose of the present study was to evaluate the systemic cardiovascular effects of GSK1016790A, a novel TRPV4 activator, and to examine its mechanism of action. In three species (mouse, rat, and dog), the i.v. administration of GSK1016790A induced a dose-dependent reduction in blood pressure, followed by profound circulatory collapse. In contrast, GSK1016790A had no acute cardiovascular effects in the TRPV4−/− null mouse. Hemodynamic analyses in the dog and rat demonstrate a profound reduction in cardiac output. However, GSK1016790A had no effect on rate or contractility in the isolated, buffer-perfused rat heart, and it produced potent endothelial-dependent relaxation of rodent-isolated vascular ring segments that were abolished by nitric-oxide synthase (NOS) inhibition (N-nitro-l-arginine methyl ester; l-NAME), ruthenium red, and endothelial NOS (eNOS) gene deletion. However, the in vivo circulatory collapse was not altered by NOS inhibition (l-NAME) or eNOS gene deletion but was associated with (concentration and time appropriate) profound vascular leakage and tissue hemorrhage in the lung, intestine, and kidney. TRPV4 immunoreactivity was localized in the endothelium and epithelium in the affected organs. GSK1016790A potently induced rapid electrophysiological and morphological changes (retraction/condensation) in cultured endothelial cells. In summary, inappropriate activation of TRPV4 produces acute circulatory collapse associated with endothelial activation/injury and failure of the pulmonary microvascular permeability barrier. It will be important to determine the role of TRPV4 in disorders associated with edema and microvascular congestion.
Evidence suggests that the transient receptor potential (TRP) vanilloid subtype 4 (V4), a member of the TRP family, is a thermo/osmo/mechanosensitive cationic channel that regulates intracellular Ca2+-homeostasis and cell volume (for review, see Plant and Strotmann, 2007). The TRPV4 message is expressed in cardiovascular tissues (heart and blood vessels), and evidence of functional expression has been demonstrated in vascular smooth muscle and endothelial cells (Earley, 2006; Inoue et al., 2006; Yang et al., 2006). In the endothelium, activation of TRPV4 by ligands or shear-stress triggers nitric oxide (NO)-dependent vasorelaxation (Kohler et al., 2006). These studies suggest that TRPV4 activation is linked mechanistically to NO generation during the process of endothelial mechanotransduction.
TRPV4 also seems to play a role in fluid distribution and integrity of endothelial/epithelial barriers. It is important to note that TRPV4 activation in the lung microvasculature increases endothelial and epithelial permeability and preferentially causes barrier disruption in alveolar septal regions (Alvarez et al., 2006). Increased paracellular permeability associated with disruption of tight junctions has also been associated with TRPV4 activation in the epithelium (Reiter et al., 2006). Most recently, it has been demonstrated that TRPV4 is a major determinant of acute microvascular injury in the lung caused by mechanical ventilation and increased lung permeability induced by elevated pulmonary vascular pressure (Hamanaka et al., 2007; Jian et al., 2008). Taken together, these results suggest that TRPV4 mechanisms affect local fluid distribution and modulate barrier integrity.
TRPV4 is expressed to some degree in the vascular smooth muscle, where activation by endogenous mediators [5,6-epoxyeicosatrienoic acid (EET) or 11,12-EET] or 4α-PDD elicits a nifedipine-insensitive inward Ca2+ current (Yang et al., 2006). In cerebral arteries, TRPV4 activation is associated with Ca2+-mediated large conductance Ca2+-activated K+ channel activation, hyperpolarization, and vasodilation (Earley, 2005). However, this scenario is probably different in the pulmonary vascular smooth muscle, where 11,12-EET activates TRPV4 and is known to cause pulmonary vasoconstriction (Yang et al., 2006).
Despite these compelling observations suggesting an important role of TRPV4 in cardiovascular homeostasis, there are no published reports describing the integrated circulatory actions of TRPV4 activation. The aim of the present study was to examine the systemic hemodynamic effects of a potent and selective TRPV4 agonist (GSK1016709A) and to probe the underlying functional mechanisms. Using standard isolated tissue and in vivo preparations, we have found that TRPV4 activation produces dramatic and complex cardiovascular effects associated with endothelial barrier failure.
Materials and Methods
All animal experiments were conducted in accordance with the Institute of Laboratory Animal Resources (1996) and approved by the GlaxoSmithKline Animal Care and Use Committee.
In Vivo Hemodynamic Studies
Hemodynamic Measurement in Anesthetized Mice.
Blood pressure and heart rate (HR) were evaluated in several mouse strains, i.e., wild-type (WT; TRPV4+/+), TRPV4+/−, TRPV4−/− (Charles River Laboratories, Inc., Wilmington, MA), B6.129P2-NOS3/J [endothelial NO synthase (eNOS)−/−], and C57BL/6 (The Jackson Laboratory, Bar Harbor, ME). Anesthesia was induced and maintained with isoflurane (1.5% in O2). The right common carotid artery and jugular vein were isolated and cannulated with polyethylene 50 cannulae for continuous monitoring of blood pressure, HR, and for infusion of drugs. GSK1016790A doses were administered in an escalating fashion at the rate of 20 μl/min in a total volume of 100 μl. The vehicle was 1% dimethyl sulfoxide (DMSO)/20% Captisol (sulfobutyl ether-β-cyclodextrin; CyDex, Lenexa, KS) in saline.
Hemodynamic Measurement in the Anesthetized Dog.
Male Marshall Beagles weighing 8 to 12 kg were fasted for 18 h and then anesthetized with propofol (10 mg/kg i.v.). A tracheotomy was performed, and dogs were ventilated mechanically with room air. A Millar microtip transducer (Millar Instruments, Inc., Houston, TX) was placed into the left ventricle via the left carotid artery to monitor left ventricular pressure and its first derivative (dP/dt). A Swan-Ganz catheter (Edwards LifeScience, Irvine, CA) was placed into the pulmonary artery via the right jugular vein. Position was verified by balloon inflation and demonstration of pulmonary wedge pressure. Femoral arteries and veins were isolated, and an arterial catheter was inserted into the left femoral artery to monitor blood pressure. Catheters were inserted into femoral veins for drug and anesthesia administration. A left thoracotomy was performed at the third intercostal space, and a 16-mm Transonic flow probe was placed around the ascending aorta to measure cardiac output. Blood gas measurements were routinely performed and maintained within normal limits until administration of GSK1016790A. Anesthesia was maintained with alpha chloralose (65 mg/kg i.v. + 0.5 mg/kg/min). All parameters were continuously recorded on CA Recorder computer software (DISS, LLC, Pinckney, MI). Dogs were allowed to equilibrate for 30 to 60 min before drug administration. GSK1016790A was administered as an ascending i.v. infusion, and each dose, beginning with vehicle, was infused for 15 min. Some dogs were administered the autonomic ganglia blocker hexamethonium chloride (5 mg/kg i.v. followed by a 100 μg/kg/min infusion) before administration of GSK1016790A. The vasopressor response induced by temporarily occluding the right common carotid artery recorded before and after hexamethonium chloride administration to verify ganglionic blockade.
Evans Blue Distribution, Histology, and TRPV4 Immunohistochemistry
Vascular permeability was assessed grossly as described previously (Sulpizio et al., 2005). In brief, rats and mice were anesthetized as described above, and catheters were placed in the carotid artery and jugular vein for the direct measurement of blood pressure and drug administration, respectively. After a 10-min equilibrium period, animals received an i.v. bolus (2 ml/kg) of Evans blue dye (15 mg/ml, containing heparin 33 units/ml) followed by infusion of GSK1016790A (30 μg/ml at 0.1 ml/min). In the low-dose group, the infusion of GSK1016790A was discontinued when the blood pressure (BP) was reduced 20 mm Hg. In the high-dose group, the infusion was terminated when the BP was reduced >50 mm Hg. The approximate dose in the low- and high-dose groups was 30 μg/kg and 60 μg/kg, respectively. Immediately after drug infusion, the thoracic cavity was opened by a single midline incision, the animal was exsanguinated by cutting open the right atrium, and the left ventricle was perfused with normal saline at 100 mm Hg until the perfusate was clear. The tissue distribution of Evans blue was assessed grossly and photographed during the postmortem examination. Evans blue dye was purchased from Sigma-Aldrich (St. Louis, MO).
Histologic assessment of tissues was performed in four groups of rats (n = 3–4 per group). One group received a vehicle infusion, and three groups received GSK1016790A (30 μg/ml at 0.1 ml/min). GSK1016790A groups were dosed to effect, i.e., a decrease in BP by 20 mm Hg, 50 mm Hg, and ≥70 mm Hg (circulatory collapse). Organs were rapidly removed, sliced, and immersed in 10% buffered formalin overnight. Histologic sections prepared from the liver, kidney, aorta, heart, lung, colon, cecum, small intestine, and mesentery were examined independently by board certified veterinary pathologists (H.T. and C.E.F.). The tissues were embedded in paraffin wax and sectioned at 5 μm followed by hematoxylin and eosin staining.
Immunohistochemistry
Twelve-week-old Sprague-Dawley rats (Charles River Laboratories, Inc.) were euthanized, and heart, liver, brain, small intestine, kidney, and lung tissues were fixed by immersion in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) and processed to paraffin. A polyclonal antibody specific for a synthetic peptide identical to the first 16 amino acids of rat TRPV4, MADPGDGPRAAPGDVA, was prepared in rabbits and affinity-purified (Invitrogen, Carlsbad, CA). Eight-micron-thick paraffin sections were dewaxed and subjected to antigen retrieval. Antigen retrieval was performed by a steam heat method with Citra Solution buffer (Biogenex, San Ramon, CA) at 95°C for 15 min. The sections were sequentially blocked for endogenous biotin binding using the Vector blocking kit (Vector Laboratories, Burlingame, CA), endogenous peroxidase activity with a 1% hydrogen peroxide, 0.2 M sodium azide solution in phosphate-buffered saline, and then with 10% normal goat serum (Jackson ImmunoResearch Inc., West Grove, PA) in staining buffer. The TRPV4-specific antibody was incubated on the sections at a concentration of 2.0 μg/ml overnight at room temperature. A purified rabbit IgG (Zymed Laboratories, South San Francisco, CA; Invitrogen) was used at the same concentration as a negative control. Bound TRPV4 antibody was detected by the standard avidin-biotin indirect peroxidase immunohistochemical technique using a biotinylated rabbit anti-rat IgG (Vector Laboratories) in combination with the Vector Elite ABC system (Vector Laboratories). Staining was visualized with 3,3′-diaminobenzidine tetrahydrochloride substrate (Dako North America, Inc., Carpinteria, CA), and the sections were then counterstained with Harris' hematoxylin (Thermo Fisher Scientific, Waltham, MA). Images were acquired using a Nikon E80i microscope (Nikon, Tokyo, Japan), a Nikon DXM 1200C camera (Optical Apparatus Inc., Ardmore, PA), and Nikon Elements BR imaging program (Nikon).
In Vitro Vascular and Cardiac Studies
Mouse and Rat Aortic Ring Segments.
Vascular reactivity studies were performed as described previously (Behm et al., 2003). Male eNOS knock-out mice (and corresponding wild-type littermates) and female Sprague-Dawley rats were euthanized via cervical dislocation (while anesthetized with 5% isoflurane in O2) or CO2 asphyxiation, respectively. The thoracic aorta was isolated, cleaned of adherent tissue, and cut into 3-mm rings. Each ring was suspended in organ baths containing Krebs' solution of the following composition: 112 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, 25 mM NaHCO3, and 11 mM dextrose. Krebs' solution was maintained at 37°C and aerated with 95% O2/5% CO2, pH 7.4. Standard force displacement transducers and recording apparatus (MLT0201/D; Letica Scientific Instruments, Barcelona, Spain; ADInstruments, Colorado Springs, CO; TSD125; BIOPAC Systems, Inc., Goleta, CA) were used to measure changes in aortic isometric force. Resting tension for vascular ring segments in the mouse was set to 0.5 g and 1.0 g in the rat. In some rat aortic segments, the endothelium was removed by passing an 18-gauge, 1.5-inch hypodermic needle through the lumen of the ring and gently rubbing the interior of the vessel against the needle shaft. Once mounted in the tissue bath, the arteries were allowed to equilibrate for 45 to 60 min, and viability was assessed by recording and washing out responses to 60 mM KCl and 100 nM phenylephrine. The functional integrity of the aortic endothelium was determined by evaluation of carbachol-induced (10 μM mouse; 1 μM rat) relaxation in vessels precontracted with phenylephrine (100 nM). After washout, tissues were again precontracted with an EC80 concentration of phenylephrine (100 nM), and single point or concentration-response experiments were performed to assess GSK1016790A-induced vasorelaxation. DMSO (10 μl) was used as a vehicle/time control. Some endothelium intact rat aorta segments were pretreated for 30 min with l-NAME (100 μM) before contraction with phenylephrine. All responses were allowed to reach steady state before changes in concentration were made. Data are expressed as the mean percentage change in the phenylephrine-induced contraction ±S.E.M.
Langendorff-Perfused Heart.
As described previously (Yue et al., 2000), male Sprague-Dawley rats (∼350 g) were anesthetized with an i.p. injection of Nembutal (60 mg/kg) and heparinized with sodium heparin 200 (IU/kg). The hearts were rapidly excised and transferred in ice-cold buffered Krebs-Henseleit solution, consisting of 118.5 mM NaCl, 4.7 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 25 mM NaHCO3, 1.2 mM KH2PO4, and 11 mM glucose, bubbled with 95% O2, 5% CO2, pH 7.4. A retrograde aortic cannula was immediately placed and connected to the Langendorff apparatus [Isolated Heart Size 1 (IH-1); Harvard Apparatus Inc., Holliston, MA] to start buffer perfusion at a constant pressure of 70 mm Hg (37°C). A water-filled latex balloon, connected to a blood pressure transducer, was inserted through the mitral valve into the left ventricle. The balloon was inflated with water until the left ventricular end-diastolic pressure was 5 to 8 mm Hg, and isovolumic left ventricular-developed pressures were recorded to a digital data acquisition system. Vehicle and drug were added to the perfusion buffer.
Cell Cultures and Morphological Assessment.
Human umbilical vein endothelial cells (HUVECs) and human aortic smooth muscle cells (AoSMCs) were obtained from Lonza Walkersville, Inc. (Walkersville, MD). HUVECs were grown in endothelial basal medium supplemented with growth factors and 2% fetal bovine serum (Lonza Walkersville, Inc.), and AoSMCs were grown in Dulbecco's modified eagle's medium supplemented with 10% fetal bovine serum in a humidified environment of 5% CO2, 95% air at 37°C. Cells were grown in 2-well chamber slides (Nalge Nunc International, Rochester, NY) and used at a subconfluent density. Before experiments, the medium was changed to basal medium containing 2% fetal bovine serum for HUVECs and AoSMCs.
Cells treated with vehicle or GSK1016790A for the indicated time intervals were gently washed with phosphate-buffered saline, fixed with 4% paraformaldehyde, and stained with Giemsa stain (Sigma-Aldrich). For cell imaging, HUVECs were fixed, permeabilized, and stained with mouse anti-human α-tubulin antibody (1:100) followed by goat anti-mouse IgG conjugated with Alexa Fluor 488 (1:500) (Invitrogen).
To quantify the cells attachment after GSK1016790A treatment, 10 randomly selected fields from each slide were counted double blinded. Cell counts from the vehicle treatment were normalized as 100%.
Patch-Clamp Analysis
Human aortic endothelial cells (HAECs) were purchased from Lonza Walkersville, Inc., and cultured according to the supplier's instructions. Whole-cell patch-clamp experiments were performed on HAECs at passage 5. Experimental conditions were similar to what have been described previously (Zeng et al., 2006). Patch-clamp pipette solution was as follows: 140 mM KCl, 5 mM EGTA, 1 mM MgCl2, 5 mM MgATP, 0.2 mM CaCl2, and 5 mM HEPES, pH 7.2. External solution was as follows: 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4. The membrane current was elicited in HAECs using a ramp voltage-clamp protocol repeated every 10 s (holding potential = −80 mV, depolarizing from −100 mV to +80 mV in 700 ms) at room temperature.
Drugs and Formulations
The TRPV4 agonist, GSK1016790A (shown in Fig. 1), was synthesized in the Department of Medicinal Chemistry of the Oncology Center of Excellence in Drug Discovery, GlaxoSmithKline Pharmaceuticals (Upper Providence, PA) for the purpose of investigating the biology of the TRPV4 channel in a variety of systems. GSK1016790A for in vivo administration was prepared in a vehicle of 1% DMSO/20% sulfobutyl ether-β-cyclodextrin (Captisol; CyDex) in saline and in DMSO (0.1%) for in vitro studies.

A, the chemical structure and in vitro characterization of GSK1016790A in recombinant cellular assays and across species in native cells. B, GSK1016790A activates a cationic current in primary cultured human aorta endothelial cells. Top, time course of the membrane current measured at +80 mV and −95 mV. Horizontal bars indicate external drug application. Bottom, current/voltage relationship in control, 10 nM GSK1016790A, and 10 nM GSK1016790A in the presence of 5 μM RuR.
Statistical Analysis
Data are presented as mean ± S.E.M. Differences between groups were compared by paired and unpaired Student's t tests or by analysis of variance followed by a Bonferroni test. P values of <0.05 were considered statistically significant.
Results
Characterization of GSK1016790A, a TRPV4 Activator.
GSK1016790A is a recently described novel chemical entity that potently activates TRPV4 channels in a variety of recombinant and native cellular assays (Fig. 1A). GSK1016790A has single-digit nanomolar potency in human, dog, and bovine cellular assays (1.0–5.0 nM) and is slightly less potent in rodent assays (10–18.5 nM). No activity is detectable versus the highly homologous TRP vanilloid subtype 1 (V1) cation channel, as assessed by Ca2+ influx studies in TRPV1-transduced cells (see Supplemental Fig. 2). The addition of GSK1016970A (10 nM) to the recording chamber (exterior of the cell) activated both inward and outward currents in HAECs (Fig. 1B). The activated current showed some outward rectification and had a reversal potential near 0 mV. The GSK1016970A-activated current was largely inhibited by ruthenium red (RuR; 5 μM), a nonspecific TRP channel blocker. GSK1016790A had approximately 1000-fold greater potency than the standard TRPV4 activator, 4α-PDD (data not shown).
Hemodynamic Studies.
In wild-type mice (TRPV+/+), the i.v. administration of GSK1016790A induced a dose-dependent reduction in blood pressure that culminates in cardiovascular collapse at the highest dose (Fig. 2A). The threshold dose was approximately 0.03 mg/kg (data not shown), the depressor dose was 0.1 mg/kg, and the lethal dose was 0.3 mg/kg (n = 3). The TRPV4+/− heterozygotic strain exhibited reduced sensitivity to the blood pressure effects of GSK1016790A (Fig. 2C)—the depressor dose was 0.3 mg/kg, and the lethal dose was 1.0 mg/kg (n = 3). TRPV4 gene deletion (TRPV4−/−) abolished the hemodynamic effects of GSK1016790A (Fig. 2D). Vehicle administration had no significant effect on BP.

Typical tracings illustrate the effects of GSK1016790A on MAP in wild-type (A, TRPV4+/+) and congenic mouse strains (C, TRPV4+/−; D, TRPV4−/−; B, eNOS−/−). D, vasodepression and circulatory collapse were observed in all strains, with the exception of TRPV4−/−. All doses (mg/kg) were administered at arrows in a volume of 100 μl (20 μl/min).
GSK1016790A was examined in greater detail in the anesthetized dog to determine the hemodynamic events responsible for TRPV4-mediated circulatory collapse. Similar to the mouse, the i.v. administration of GSK1016790A caused a reduction in blood pressure and eventual circulatory failure in the dog. At the threshold dose (0.3 μg/kg/min), modest reductions in mean arterial pressure (MAP) and systemic vascular resistance (SVR) were observed (Fig. 3). The circulatory collapse observed at 1.0 μg/kg/min was clearly related to acute cardiac output failure determined largely by a reduction in stroke volume. Pretreatment with the ganglionic blocker, hexamethonium, did not alter the overall pattern of the hemodynamic response; however, the magnitude of the collapse was exaggerated. GSK1016790A had similar hemodynamic effects in the rat.

The effect of 15-min i.v. infusions of vehicle (V) or GSK1016790A (0.1, 0.3, and 1.0 μg/kg/min) on MAP (A), HR (B), cardiac output (CO; C), and SVR (D) in the presence or absence of ganglion-blocking doses of hexamethonium (Hex) in chloralose-anesthetized dogs (n = 4/group).
Isolated Perfused Rat Heart.
To investigate the direct cardiac effects of TRPV4 activation, GSK1016790A was examined in isolated buffer-perfused rat hearts, where left ventricular function was monitored by recording developed pressure in a balloon-tipped catheter inserted in the left ventricle. GSK1016790A (1.5–13.5 μM) in the perfusate had no effect on spontaneous rate or contractility in the isolated-perfused rat heart (Table 1).
The effect of GSK1016790A in the isolated perfused rat heart
All values are expressed as mean percentage change in baseline; n = 3 per group.
Vascular Reactivity and Role of eNOS in Circulatory Collapse.
Direct vascular effects of GSK1016790A were investigated in the rat and mouse by recording isometric tension from isolated aortic ring segments suspended in organ baths containing Krebs-Henseleit buffer. The addition of GSK1016790A to the organ bath had no effect on resting tension in any of the preparations (data not shown), but it caused potent and efficacious relaxation of vessels precontracted with phenylephrine (Fig. 4, A and B). In Fig. 4A, maximal relaxation induced by GSK1016790A was abolished by endothelial denudation, NOS inhibition (l-NAME), and the TRP channel blocker RuR. Likewise, GSK1016790A-induced relaxation of the thoracic aorta was not observed in mice lacking eNOS (Fig. 4B). However, circulatory collapse induced by GSK1016790A (n = 3) was unaltered in the eNOS−/− strain (Fig. 4B).
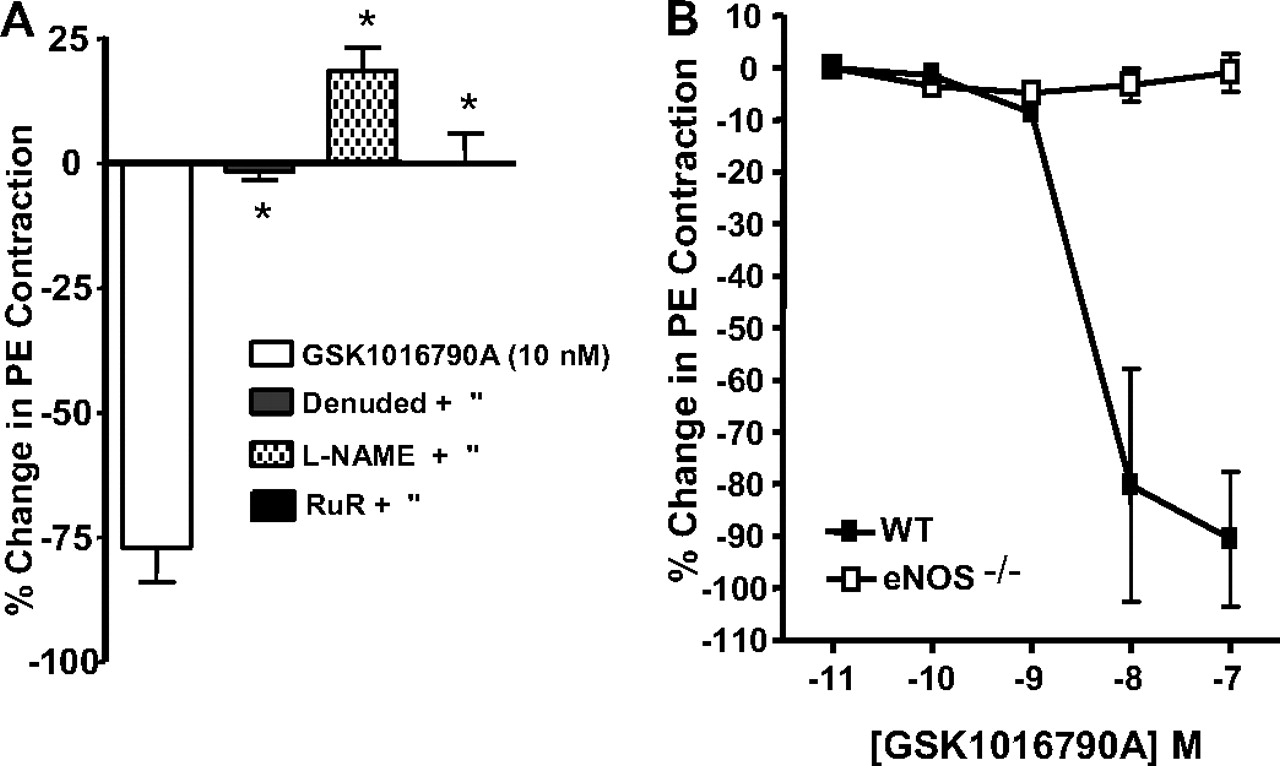
Relaxation induced by GSK1016790A in rat aortic rings from endothelium intact, endothelium-denuded, and l-NAME (100 μM) treated groups (A) and in aortic rings prepared from wild-type and eNOS null mice (B). Rat and mouse preparations were precontracted with phenylephrine (100 nM). Typical tracing of the mean arterial blood pressure effect of GSK1016790A in eNOS null mice (n = 3). The values in A and B are mean ± S.E. *, p < 0.01 versus GSK1016790A (n = 4–8/group).
Vascular Permeability.
Cardiac MRI and the tissue distribution of Evans blue were examined grossly in rats following the administration of GSK1016790A at vasodepressor doses and lethal doses. At the lethal dose, cardiac magnetic resonance imaging (MRI) (Fig. 5A) indicates signs of acute right ventricular failure characterized by right ventricular dilation with septal shift (Fig. 5B) that was accompanied by elevated pulmonary arterial pressure (Fig. 5C). GSK1016790A elicited dose-dependent extravasation of Evans blue and/or obvious vascular hemorrhage, within minutes of administration in the lung (Fig. 5, D and E), large intestine, and kidney (data not shown) at the nadir of the blood pressure response. At 15 min after GSK1016790A (0.3 mg/kg/15 min), the lung wet weight to body weight ratio was increased approximately 2.5-fold compared with vehicle [0.2 ml of DMSO (1%), 29% β-cyclodextrin] (Fig. 5F). The profound effects of GSK101670A in the lung were associated with histologic changes that included evidence of pulmonary edema and congestion associated with perivascular hemorrhage and multifocal alveolar hemorrhage (Fig. 5, G and H). At a dose (30–100 μg/kg i.v.) causing a 20 mm Hg reduction in blood pressure, there were no drug-related abnormalities detected, i.e., similar to vehicle-infused animals (data not shown). Significant histologic changes were also observed in the large intestine and included dose-dependent submucosal and mucosal edema ranging in severity from minimal to moderate (depicted in Supplemental Fig. 1). In the kidney, there was a dose-dependent acute medullary tubular necrosis that ranged in severity from minimal to moderate. In addition, there was evidence of tubular and peritubular hemorrhage. It is notable that Evans blue extravasation and hemorrhage were not observed grossly in the brain, heart, liver, pancreas, or skeletal muscle, and there was no evidence of hemorrhage in the other tissues examined histologically. Similar results were observed in wild-type mice but not in the TRPV4−/− strain.

Representative results of the TRPV4 activator, GSK1016790A, in the anesthetized Sprague-Dawley rat. End-diastolic cardiac MRI after i.v. vehicle infusion at 0.1 ml/min (A) or infusion of GSK1016790A over a dose-range (50–113 μg/kg i.v.) sufficient to induce circulatory collapse (B). C, changes in pulmonary arterial pressure observed after the infusion of GSK1016790A (arrow). D, Evans blue extravasation in the lung after vehicle, or GSK1016790A administration sufficient to cause circulatory collapse (E). F, the lung weight to body weight ratio (LW/BW) was determine 15 min after the administration of vehicle (1% DMSO + 29% β-cyclodextrin) or GSK1016790A (0.3 mg/kg/min). G and H, representative pulmonary histologic findings after the administration of vehicle and GSK1016790A, respectively (H). All observations represent 3 to 5 replicates.
Immunohistochemical Localization of TRPV4.
Immunohistochemical localization of TRPV4 was examined in a variety of rat tissues to investigate expression patterns that may underlie the heterogeneous effects of GSK101670A on microvascular permeability. A generalized pattern of immunoreactive TRPV4 staining was identified in endothelium and epithelium. In the lung, robust TRPV4 staining was observed in epithelium lining secondary and tertiary segments of the airway and in bronchial epithelium and pulmonary artery endothelium (Fig. 6, A and C). Large arteries also exhibited a more variable pattern of TRPV4 staining in the adventitium. A lacey pattern of TPV4 staining was also evident in alveolar septal regions. Vascular and bronchial smooth muscle were devoid of TRPV4 immunoreactivity.

Immunohistochemical localization of TRPV4 in normal rat lung (A–C), small intestine (D and E), and kidney (F and G); immunoperoxidase staining with hematoxylin counterstaining. TRPV4-positive structures (brown) were morphologically consistent epithelium and endothelium in respective organs. B, a preimmune rabbit IgG-negative control. ASR, alveolar septal region; ATL, ascending thin limb; Br, bronchiole; CT, convoluted tubule; EC, endothelium; PA, pulmonary artery; PAr, pulmonary arteriole; SBV, submucosal blood vessel; SM, submucosal smooth muscle; VSM, vascular smooth muscle. All scale bars equal 15 μM.
In the kidney, TRPV4-immunoreactive staining patterns were morphologically consistent with a previous description (Tian et al., 2004), i.e., robust TRPV4 staining in the nephron epithelium included ascending thick limb, distal convoluted tubule, ascending thin limb, and inner medullary collecting duct (Fig. 6, F and G). The renal arcuate artery endothelium was consistently positive for TRPV4 immunoreactivity. Arterial smooth muscle, proximal tubular segments, and glomeruli were negative. A similar general pattern of endothelial and epithelial TRPV4 immunoreactivity without smooth muscle staining was observed in the small intestine (Fig. 6, D and E).
Blood vessels throughout the brain and heart also exhibited highly restricted endothelial TRPV4 immunoreactivity (Fig. 7, A, B, and D). With the exception of the choroid in cerebral ventricles (Fig. 7C), which was strongly positive (as described by Liedtke et al., 2000), there was little or no extravascular TRPV4 immunoreactivity in the heart and only highly restricted circumventricular staining in the brain (data not shown).

Immunohistochemical localization of TRPV4 in normal rat heart (A and B) and brain (C and D) immunoperoxidase staining with hematoxylin counterstaining. TRPV4-positive structures (brown) were morphologically consistent epithelium and endothelium in respective organs. CM, cardiac muscle; CAE, cerebral arteriole endothelium; ECP, epithelium of the choroid plexus; ICA, intramural coronary arterioles; EC, endothelium. All scale bars equal 15 μM.
Endothelial and Vascular Smooth Muscle Cell Culture.
The cellular events underlying microvascular damage induced by TRPV4 activation were investigated in cultures of HUVECs and AoSMCs. The addition of GSK1016790A to culture media caused a potent (10 nM) and rapid (within 10 min) retraction and condensation of HUVECs (Fig. 8, A and B). More prolonged incubation (30 min) with GSK1016790A caused a concentration-related detachment of HUVECs from the plate (Fig. 8, G and H). The effects of GSK1016790A on HUVECs were abolished by pretreatment with the TRP channel blocker RuR (Fig. 8, C and D). In contrast to the HUVECs, GSK1016790A (100 nM) had no effect on morphology or adhesion in AoSMCs (Fig. 8, E and F) or human small airway epithelial cell cultures (data not shown). The results suggest that failure of the endothelial permeability barrier induced by TRPV4 activation is mediated by direct effects on endothelial cell morphology.

Effect of TRPV4 activator GSK1016790A on HUVECs (A–D) and AoSMCs (E and F). Cells were treated with vehicle (0.1% DMSO) (A and E), GSK1016790A (B and F), RuR alone (C), or RuR with GSK1016790A (D) at the concentrations indicated. The inserts in A and B were HUVECs stained with anti-α-tubulin. Rightward panels depict the time (H) and concentration (G) dependent effects of GSK1016790A on HUVEC attachment to the plate in the absence or presence of 1 μM RuR. H, HUVECs were preincubated with RuR for 20 min before the addition of GSK1016790A (10 nM) for 3 h. The values are mean ± S.E. **, p < 0.001 versus the vehicle.
Discussion
In the present study, we characterized a novel synthetic TRPV4 channel activator, GSK1016790A, and examined its cardiovascular effects. We provide evidence supporting the role of TRPV4 in Ca2+-dependent regulation of endothelial NO production and describe for the first time catastrophic cardiovascular effects associated with exogenous activation of TRPV4. Moreover, we provide evidence that circulatory collapse induced by TRPV4 activation is mediated by a NO-independent failure of the endothelial-epithelial permeability barrier in the lung and other selected tissues.
GSK1016790A is a novel and potent small molecule activator of the TRPV4 channel. It is structurally unrelated to phorbol ester TRPV4 activators and is inactive at highly homologous TRPV1 channels. In addition, GSK1016790A retains nanomolar potency at its target across a variety of species, making it an attractive tool for investigating the pharmacology and physiology of TRPV4 channels.
In vivo, the i.v. administration of GSK1016790A induced a dose-related depression of blood pressure followed by circulatory collapse in WT mice, rats, and dogs. In contrast, GSK1016790A had no effect on BP or HR in TRPV4−/− null mice at doses >10-fold the lethal dose in WT mice, and heterozygous TRPV4+/− mice were slightly more resistant to the lethal effects of GSK1016790A (approximately 3-fold the lethal dose in WT). These results clearly demonstrate the role of TRPV4 in mediating the cardiovascular effects of GSK1016790A. It is noteworthy that TRPV4-deficient mice were normotensive (data not shown). Hemodynamic analysis of GSK1016790A in mechanically ventilated anesthetized dogs was characterized by a reduction in cardiac output associated primarily with decreased stroke volume and delayed bradycardia. To eliminate reflex adjustments and focus on the primary actions of GSK1016790A, hemodynamic effects were examined in dogs after autonomic blockade with hexamethonium. Under these conditions, the hemodynamic actions of GSK1016790A were qualitatively similar, albeit greater in magnitude, compared with the intact preparation, i.e., a slight decrease in SVR associated with a moderate reduction in BP that was rapidly followed by the cardiac failure described above. These in vivo results suggest that GSK1016790A severely depressed cardiac function and only modestly reduced vascular tone.
Evidence suggests that the vascular effects of TRPV4 activation are, in some instances, mediated by endothelial NO generation (Kohler et al., 2006). As such, we evaluated the effects of pharmacologic and genetic manipulation of eNOS on the vascular and hemodynamic effects of GSK1016790A. In preconstricted mouse and rat-isolated aortae, GSK1016790A induced potent and efficacious relaxation. The actions of GSK1016790A in the aorta were absent in vessels obtained from TRPV4 null mouse and abolished by the nonselective TRP channel blocker, ruthenium red. These results suggest that the vascular relaxation induced by GSK1016790A was mediated specifically by activation of the TRPV4 channel. Furthermore, aortic relaxation induced by GSK1016790A was absent in eNOS null mice and abolished by endothelial denudation and inhibition of nitric-oxide synthase (l-NAME). The present results are consistent with previously described expression of TRPV4 in endothelial cells (Yao and Garland, 2005; Kwan et al., 2007) and with a recent report describing endothelial/NO-dependent relaxation of isolated rat carotid artery with a selective TRPV4 activator, 4α-phorbol-12,13-didecanoate (Kohler et al., 2006). Taken together, the results suggest that potent exogenous activation of TRPV4 is a powerful endothelial-dependent mechanism, regulating vascular tone via eNOS activation and NO generation.
Hemodynamic mechanisms of GSK1016790A were further examined in vivo in eNOS null mice and l-NAME-treated rats. Somewhat surprisingly, the hemodynamic effects and lethal dose of GSK1016790A were indistinguishable in eNOS null and WT mice. Likewise, l-NAME treatment failed to alter the circulatory collapse in rats. Thus, the TRPV4-mediated endothelial NO generation, which played a critical role in GSK1016790A-induced vasorelaxation, seems to play little or no role in TRPV4-mediated circulatory failure.
In addition, GSK1016790A had no effects on inotropy or chronotropy in the isolated rat heart. Again, the results were surprising given the profound in vivo effects of GSK1016790A on cardiac output; however, the lack of effect is consistent with the paucity of TRPV4 expression in cardiomyocytes demonstrated by TRPV4 immunohistochemistry.
Recent evidence suggests that TRPV4 activation, whether by exogenous activators or by elevated vascular and/or inspiratory pressure, induces a heterogeneous increase in pulmonary endothelial permeability (Alvarez et al., 2006; Hamanaka et al., 2007). In the present study, GSK1016790A caused a dose-dependent increase in microvascular permeability (Evans blue leakage) and hemorrhage in the lung, kidney, and intestine. The microvascular injury in the lung was associated with histologic changes that included pulmonary edema and congestion associated with perivascular hemorrhage and multifocal alveolar hemorrhage and was related to the magnitude of the hemodynamic response. The lung injury was associated with pulmonary hypertension and right heart failure that most likely obscure observation of NO-related hemodynamic effects. It is noteworthy that in the present study, evidence of gross microvascular injury was not apparent in the heart, brain, liver, or skeletal muscle, suggesting a heterogeneous microvascular response to TRPV4 activation.
The present results provide evidence for localization and function of TRPV4 channels in vascular endothelium that is consistent with a role in the regulation of vascular permeability. However, the data do not provide a clear explanation for the organ-specific changes in vascular permeability observed after TRPV4 activation in vivo. A paucity of TRPV4 channels in resistant organs, e.g., heart and brain, can not be strictly ruled out, but it seems highly unlikely given the robust endothelial TRPV4 immunoreactivity in these organs. One commonality in the sensitive organs (lung, intestine, and kidney) is the unique juxtaposition of TRPV4-positive epithelium and endothelium. Thus, it is tempting to speculate that in vivo activation of TRPV4 in both of these cell types plays a cooperative role in mediating the effects of GSK1016790A on vascular permeability. Temporal considerations and cell-specific TRPV4 signaling are also areas for future study.
At the cellular level, GSK1016790A caused apparent cytoskeletal reorganization characterized by rapid endothelial cell retraction/contraction, condensation, and subsequent detachment. All morphologic changes in endothelial cells were inhibited by ruthenium red. These results are consistent with the complex interactions of eNOS, TRPs, and the cytoskeleton that are known to exist at endothelial caveoli microdomains and are analogous to actions attributed to TRPC4 (Govers and Rabelink, 2001; Tiruppathi et al., 2006). In contrast to endothelial cells, the morphology of vascular smooth muscle cells and small airway epithelial cells (data not shown) was not affected by GSK1016790A. In addition, GSK1016790A did not contract isolated rat pulmonary artery (data not shown). The results suggest that TRPV4 activation causes disruption of the microvascular permeability barrier by directly altering endothelial morphology and integrity.
In conclusion, a novel TRPV4 activator was used to describe, for the first time, TRPV4-dependent circulatory collapse. Failure of the cardiac output was not related to TRPV4-dependent endothelial NO generation or direct myocardial actions. Evidence suggests that the circulatory collapse is the result of profound disruption of the endothelial permeability barrier in the lung and attendant acute pulmonary hypertension and right heart failure. Given the powerful effects of TRPV4 on microvascular integrity, it will be important to further define its role in endothelial function and vascular disease.
Acknowlegdments
Special thanks to Thomas Covatta for preparation of photomicrographs and to Dr. Kendal Frazier for histopathology consultation. All authors are employed by GlaxoSmithKline Pharmaceuticals.
Footnotes
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.107.134551.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- TRP
- transient receptor potential
- V4
- vanilloid subtype 4
- NO
- nitric oxide
- EET
- epoxyeicosatrienoic acid
- HR
- heart rate
- WT
- wild type
- eNOS
- endothelial NO synthase
- DMSO
- dimethyl sulfoxide
- BP
- blood pressure
- l-NAME
- N-nitro-l-arginine methyl ester
- HUVEC
- human umbilical vein endothelial cell
- AoSMC
- human aortic smooth muscle cell
- HAEC
- human aortic endothelial cell
- V1
- vanilloid subtype 1
- RuR
- ruthenium red
- MAP
- mean arterial pressure
- SVR
- systemic vascular resistance
- MRI
- magnetic resonance imaging
- 4α-PDD
- 4α-phorbol 12,13-didecanoate.

- Received November 20, 2007.
- Accepted May 6, 2008.
- Copyright © 2008 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}