


Abstract
We have investigated the bronchodilator and anti-inflammatory properties of roflumilast (3-cyclopropylmethoxy-4-difluoromethoxy-N-[3,5-dichloropyrid-4-yl]-benzamide), a novel, highly potent, and selective phosphodiesterase 4 (PDE4) inhibitor. Additionally, we compared the effects of roflumilast and itsN-oxide, the primary metabolite in vivo, with those of the PDE4 inhibitors piclamilast, rolipram, and cilomilast. Roflumilast inhibited the ovalbumin-evoked contractions of tracheal chains prepared from sensitized guinea pigs (EC50 = 2 × 10−7 M) but showed no relaxant effect on tissues contracted spontaneously. In spasmogen-challenged rats and guinea pigs, intravenously administered roflumilast displayed bronchodilatory activity (ED50 = 4.4 and 7.1 μmol/kg, respectively). Furthermore, roflumilast dose dependently attenuated allergen-induced bronchoconstriction in guinea pigs (ED50 = 0.1 μmol/kg i.v.). Roflumilast given orally (ED50 = 1.5 μmol/kg) showed equal potency to its N-oxide (ED50 = 1.0 μmol/kg) but was superior to piclamilast (ED50 = 8.3 μmol/kg), rolipram (ED50 = 32.5 μmol/kg), and cilomilast (ED50 = 52.2 μmol/kg) in suppressing allergen-induced early airway reactions. To assess the anti-inflammatory potential of orally administered roflumilast, antigen-induced cell infiltration, total protein, and TNFα concentration in bronchoalveolar lavage fluid of Brown Norway rats were determined. Roflumilast and its N-oxide equally inhibited eosinophilia (ED50 = 2.7 and 2.5 μmol/kg, respectively), whereas the reference inhibitors displayed lower potency (ED50 = 17–106 μmol/kg). Besides, orally administered roflumilast abrogated LPS-induced circulating TNFα in the rat (ED50 = 0.3 μmol/kg), an effect shared by its N-oxide, with both molecules exhibiting 8-, 25-, and 310-fold superiority to piclamilast, rolipram, and cilomilast, respectively. These results, coupled with the in vitro effects of roflumilast on inflammatory cells, suggest that roflumilast represents a potential new drug for the treatment of asthma and chronic obstructive pulmonary disease.
Among the world's most prevalent diseases are asthma and COPD. Asthma is a chronic disease characterized by inflammation of the airways that is central to the airway dysfunction. Typically, the airway wall is infiltrated by a variety of inflammatory cells such as eosinophils, mast cells, and CD4+ T lymphocytes (type 2 T helper lymphocytes) that release a plethora of mediators ultimately causing the symptoms and the histopathology of asthma. The prevalence and severity of allergic asthma have been steadily increasing over the past 20 years together with the number of reported cases of fatal asthma, and it affects up to 10% of the population of most developed countries (O'Byrne and Postma, 1999; Giembycz, 2000). In comparison, COPD is the sixth cause of death in the world and affects 4 to 6% of people more than 45 years of age. Like asthma, COPD is characterized by airway obstruction being the conditio sine qua non of both diseases, and progressive lung inflammation that is associated with the influx of inflammatory cells, in COPD predominantly being neutrophils, macrophages, and CD8+ T lymphocytes (O'Byrne and Postma, 1999). The disease statistics clearly demonstrate the increasing need for drugs targeting the mechanisms involved in eosinophil and neutrophil activation and accumulation for the management of asthma and COPD. Glucocorticosteroids are the only drugs currently available that effectively reduce airway inflammation in asthma (Barnes and Pedersen, 1993). However, they lack selectivity, direct bronchodilatory activity, and efficacy in long-term treatment of COPD (O'Byrne and Postma, 1999). Therefore, new anti-inflammatory drugs for the treatment of asthma and COPD are required.
Potential therapeutic agents that exhibit broad anti-inflammatory and immunomodulatory activity are inhibitors of cAMP-specific phosphodiesterases (PDE). PDE4 is the predominant family of PDEs expressed in inflammatory cells, including eosinophils (Dent et al., 1991; Hatzelmann et al., 1995; Souness et al., 1995), T lymphocytes (Essayan et al., 1997), macrophages (Tenor et al., 1995; Gantner et al., 1997), neutrophils (Schudt et al., 1991), dendritic cells (Gantner et al., 1999), mast cells, and structural cells such as sensory cells and epithelial cells (Torphy, 1998). Since PDE4 metabolizes cAMP, a signal molecule known to attenuate cell activation, inhibition of PDE4 activity causes elevation of intracellular cAMP levels and subsequently down-regulation and modulation of a variety of inflammatory cell activities (Torphy, 1998; Barnette, 1999; Essayan, 1999). In animal models of asthma, PDE4 inhibitors reduce eosinophil infiltration and airway hyperresponsiveness response to allergen (for review, seeTorphy, 1998), and PDE4 has been suggested as a molecular target for a broad range of new drugs for the treatment of a variety of diseases, especially for the treatment of respiratory diseases (Barnes, 1999;Schudt et al., 1999; Torphy and Page, 2000). Several PDE4 inhibitors have been tested in humans but showed either lack of efficacy or side effects. The prototype PDE4 inhibitor rolipram caused nausea and vomiting in clinical trials (Zeller et al., 1984) and although CDP840 displayed some inhibitory effects on the late response to allergen, it is no longer in clinical development (Harbinson et al., 1997). However, encouraging data from clinical studies using cilomilast strongly support the concept that PDE4-selective inhibitors will be useful in the treatment of COPD and asthma (Barnette, 1999).
The present manuscript describes the in vivo efficacy of roflumilast, a novel orally active PDE4 inhibitor in several airway and inflammation models. Roflumilast is derived from a series of benzamides and was identified as a potent PDE4-selective inhibitor (Amschler, 1995), which is currently under clinical investigation for the treatment of COPD (phase II) and asthma (phase III). Roflumilast N-oxide is the primary metabolite of roflumilast in several animal species and humans, largely contributing to the efficacy of roflumilast in vivo (M. David, E. Sturm, and K. Zech, manuscript in preparation). The investigations to assess the pharmacological properties of roflumilast and its N-oxide metabolite were carried out in comparison to reference compounds. Piclamilast (RP 73401) was chosen for its structural proximity to roflumilast (Ashton et al., 1994), and rolipram to compare roflumilast with the archetype of PDE4-selective inhibitors. Since the second-generation PDE4 inhibitor Ariflo (cilomilast, SB 207499) is currently being investigated in clinical trials to prove efficacy in asthma and COPD, this compound served as standard for assessing the potential of roflumilast.
Experimental Procedures
Animals
Male Dunkin-Hartley guinea pigs and male Sprague-Dawley rats were purchased from Charles River/Wiga (Sulzfeld, Germany) and male BN rats were either supplied by Charles River/Wiga or the breeding institution at Byk Gulden (Konstanz, Germany). Animals were housed in groups of four to five per Macrolon cage (type IV) at 20–24°C (guinea pigs) or 20–22°C (rats) and 12-h day/night rhythm and had free access to water and food (standard diet 84525W4 for guinea pigs or maintenance diet 9439 for rats, respectively; NAFAG, Gossau, Switzerland). All animals used received humane care in accordance with national guidelines and legal regulations.
Materials
Ovalbumin (OVA; egg albumin, grade V) for the sensitization of BN rats was purchased from Fluka (Buchs, Switzerland), whereas OVA for the sensitization of guinea pigs (egg albumin, grade III), bovine serum albumin (fraction V), histamine (histamine-2HCl), hydroxylamine, indomethacin, LPS (Salmonella abortus equi), pyrilamine (pyrilamine maleate salt), and serotonin (creatine sulfate complex) were obtained from Sigma (Deisenhofen, Germany). Polyethylene glycol 400 (PEG400) and methylhydroxylpropyl cellulose (2910.15) for the preparation of the 4% methocel solution were supplied by Serva (Heidelberg, Germany) and by Dow Chemicals (Midland, MI), respectively.Bordetella pertussis vaccine was purchased from Behringwerke AG (Marburg, Germany), Diff-Quick from Baxter (Deerfield, IL), “Eosinofix” from ABX (Göppingen, Germany), heparin (Liquemin N 25000) from Hoffmann-La Roche AG (Grenzach-Wyhlen, Germany), pancuronium bromide from Organon (Eppelheim, Germany), pentobarbital (Narcoren) from Merial (Hallbergmoos, Germany), propranolol (dl-propranolol hydrochloride) from Rhein-Pharma (Plankstadt, Germany), thiopental (Trapanal) from Byk Gulden (Konstanz, Germany), urethane (ethyl carbamate) from Riedel-de Haen (Seelze, Germany), and saline (0.9% NaCl) from Braun (Melsungen, Germany). Aluminum hydroxide gel [Al(OH)3, AHG], and all other chemicals (analytical grade) were obtained from Merck (Darmstadt, Germany).
Compounds
Roflumilast (mol. wt. = 403.2), roflumilast N-oxide (mol. wt. = 419.2; both WO95/01338), piclamilast (mol. wt. = 381.3; WO92/12961), cilomilast (mol. wt. = 343.4; WO93/19749), and racemicR,S-rolipram (mol. wt. = 275.4) were synthesized by the Department of Chemistry at Byk Gulden.
For intravenous administration, a solution of roflumilast was prepared. For that, equivalent amounts of the compound were dissolved in 1.5 ml of PEG400 plus 1 ml of 0.1 N NaOH (heated to 70°C). Saline was added to a total volume of 5 ml (pH 10, 1 N NaOH). The stock solution was further diluted in saline. PEG400 (30%) in saline (pH 10) served as placebo. These solutions were slowly (within 1.5 min) injected i.v. in guinea pigs and rats at a volume of 1 ml/kg of body weight.
For oral administrations, equivalent amounts of all drugs were mixed with 0.4 ml of PEG400. This mixture was resuspended with 4% methocel solution. Control animals received the equivalent amount of the corresponding solvent p.o. at 1 h before the OVA-challenge. The p.o. administration volume was 1 ml/kg of body weight and 10 ml/kg of body weight for guinea pigs and rats, respectively, for the drugs and placebo. Since no comparative studies with roflumilast, itsN-oxide, and the reference PDE4 inhibitors were performed, it cannot be completely excluded that the vehicle methocel has an influence on plasma concentrations of these drugs. However, methocel is a vehicle that has been and is still widely used for screening of compounds from different chemical classes. While comparing some of these compounds suspended in methocel with the ones diluted in an adequate solvent, no differences in potencies of suspended or diluted orally administered inhibitors were ever observed.
Guinea Pig Isolated Trachea
Animals and Sensitization.
Male Dunkin-Hartley guinea pigs (400–450 g) were actively sensitized by i.p. injections of 1 ml/kg of body weight of a 5% (w/v) OVA (grade III; Sigma)/saline solution on days 1 and 2. Isolation of trachea was performed 3 to 4 weeks later.
Tissue Bath Experiments.
OVA-sensitized guinea pigs were killed by a sharp blow on the head and exsanguination followed by removal of the trachea. After carefully clearing off adhering tissue, the trachea was cut into single rings that were tied together forming up to six four- to five-ring chains and suspended in 10-ml organ baths containing Krebs' buffer of the following composition: 118 mM NaCl, 5.2 mM KCl, 1.9 mM CaCl2, 0.56 mM MgSO4, 0.8 mM NaH2PO4, 25.0 mM NaHCO3, and 11.1 mM glucose, maintained at 37°C and continuously aerated with a mixture of 95% O2 and 5% CO2. The ends of the chains were tied at the bottom of the tissue bath and connected to a force-displacement transducer (type K-30; Hugo Sachs Elektronik, March, Germany) for the recording of isometric tension changes, and then placed under 1.5 g of tension. After a 1-h equilibration period during which the preparations were repeatedly washed and the spontaneous contraction reached a plateau, each tracheal preparation was pretreated for 30 min with roflumilast (10−9–10−6 M),N-oxide (3 × 10−8–3 × 10−7 M), piclamilast (10−9–10−6 M), rolipram (10−7–3 × 10−6 M), and cilomilast (10−8–10−5 M) followed by cumulative administration of OVA (10−10–10−7 g/ml organ bath volume), with each OVA concentration remaining in contact with the trachea until the more sustained tonic component of the contractile response reached a plateau before addition of the next higher OVA concentration (factor 10). At the end of the OVA administration, a high concentration of carbachol (10−3 M) was additionally given to define the maximal contraction of each individual trachea. All contraction responses to OVA were expressed as percentage values of the carbachol-induced maximum response (100%). Only one concentration-response curve of OVA was generated with each tracheal chain. Results are given as means ± S.E.M. for n= 6–10 tracheal strips in each case.
Spasmogen-Induced Bronchoconstriction in the Anesthetized, Mechanically Ventilated Guinea Pig and BN Rat
Animals.
Male Dunkin-Hartley guinea pigs (400–520 g) and male BN rats (200–270 g) were used for these experiments.
Experimental Protocol, Drug Administration, and Measurement of Pulmonary Parameters.
Animals were anesthetized with urethane (1.2 g/kg i.p., 10 ml/kg). For i.v. injections, the right jugular vein was surgically exposed and cannulated with a small (outer diameter, 0.75 mm) polyethylene catheter. The trachea was exposed, sectioned, and a steel cannula (outer diameter, 2.2 mm, inner diameter, 1.9 mm) with a side port was inserted about 1.5 cm proximal to the bifurcation. This tracheal cannula was connected to a heated Fleisch pneumotachograph (0000; Hugo Sachs Elektronik), which was connected to a differential pressure transducer (Validyn DP45-14; Hugo Sachs Elektronik). The flow-signal was amplified using a carrier frequency bridge amplifier (CFBA 677; Hugo Sachs Elektronik) and fed to the data acquisition system. The side port of the cannula was attached to one port of another pressure transducer (MPX-11DP; Hugo Sachs Elektronik) to determine the pressure changes in the trachea. The signal was amplified by a DC-bridge amplifier (DBA 660; Hugo Sachs Elektronik) and sent to the data acquisition system.
About 10 min before the first histamine challenge [0.025–0.1 μmol/kg (in saline), 1-ml/kg bolus i.v. injection; 10, 20 min before and 2, 10, 30, and 60 min after drug administration] or serotonin challenge [1 μmol/kg (in 0.05% sodium metabisulfite), 1-ml/kg bolus i.v. injection; 10 and 20 min before, and 2, 10, 30, and 60 min after drug administration], the animals received pancuronium bromide (1.5 mg/kg i.v., guinea pig; 3 mg/kg i.v., BN rat; 1 ml/kg) to relax skeletal muscles and to abolish spontaneous breathing, and were then mechanically ventilated through the pneumotachograph with air using a small animal ventilator (type KTR4; Hugo Sachs Elektronik), which was adjusted to 60 breath/min, 7-ml/kg tidal volume, and 40:60% inspiration/expiration ratio (guinea pigs) or 80 breath/min, 9-ml/kg tidal volume, and 40:60% inspiration/expiration ratio (BN rat), respectively.
Solutions of the test compounds were i.v. injected as described above (see Compounds) at the time points indicated (Figs. 2 and3). The drug-free solvent served as placebo (1 ml/kg of body weight).
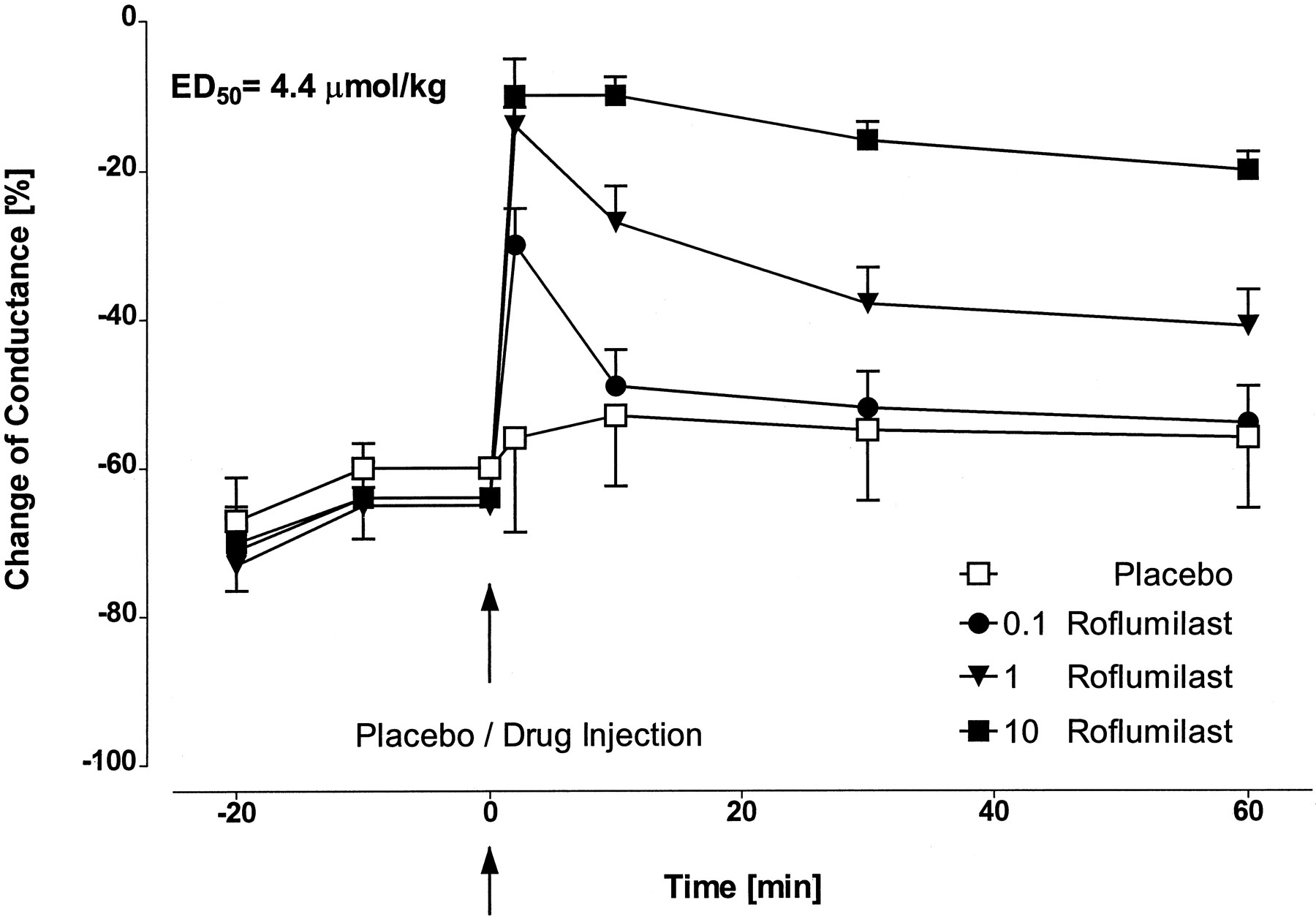
Inhibitory effect of roflumilast (0.1, 1, 10 μmol/kg i.v.) on time course of serotonin-induced decrease of airway conductance in anesthetized and ventilated BN rats. Results are given as means ± S.D.; n = 6/roflumilast dose,n = 10 for placebo.

Inhibitory effect of roflumilast (0.1, 1, 10 μmol/kg i.v.) on time course of histamine-induced decrease of airway conductance in anesthetized and ventilated guinea pigs. Results are given as means ± S.D.; n = 6/roflumilast dose and placebo.
The main lung function parameters were flow (F) measured by the pneumotachograph; inflation pressure (PIP) registered at the side port of the tracheal cannula against atmospheric pressure; and tidal volume (TV), which was integrated from the flow signal. Airway resistance (RAW) and conductance (CON = 1/RAW) were calculated from the above-given main lung function parameters according to the method described by Amdur and Mead (1958). Calculation of RAW is based on PIP and F for two isovolumetric points at 70% TV of each single breath (RAW = PIP/F cm H2O/ml/s). For acquisition, analysis and archiving of all pulmonary parameters, a commercially available PC-based software program was used (model AP1/8, version V1.10f; PO-NE-MAH Inc., distributed by Hugo Sachs Elektronik). The program is able to calculate RAW and CON, and allows averaging and searching for minimum and maximum changes at defined time intervals. The system allows to display the time courses of several primary (e.g., F, PIP) and derived parameters (e.g., TV, CON) on a monitor simultaneously and to print out the digitized values or graphs for documentation. All parameters were averaged at 1-min intervals throughout the whole experiment except for the time 0 to 60 s after each histamine or serotonin injection where averaging was performed at 4-s intervals to detect maximum and minimum changes of the pulmonary parameters.
Data Analysis.
To determine whether roflumilast altered lung mechanics, the time courses of the baseline values (=1-min averaged values before each histamine/serotonin challenge) of CON were assessed and biostatistically analyzed relative to placebo. To quantify the bronchodilatory activity of the compounds, the maximum spasmogen-induced decrease of CON was calculated in regard to the corresponding baseline values at each challenge. The maximum decrease was observed about 12 s after injection of histamine or serotonin and was in the range of 75 to 80% for the guinea pig (equivalent to 4–5-fold increase in RAW) or 50 to 75% for the BN rat (equivalent to 2–4-fold increase in RAW), respectively. As with the baseline parameters, the time course of the decrease of CON was assessed and was biostatistically analyzed relative to placebo. In case of homogeneity of the variances (Bartlett's test), a multiple Student-Welch test was used to determine the significance of the differences between the groups. If the variances were different, the paired SchefféContrasts were applied; p < 0.05 was considered statistically significant. To estimate an effective bronchodilatory dose, the inhibition of the histamine- or serotonin-induced decrease of CON was calculated relative to placebo. The values were used to calculate the ED50 based on log-linear regression analysis.
Allergen-Induced Bronchoconstriction in the Anesthetized, Mechanically Ventilated Guinea Pig
Animals and Sensitization.
Male Dunkin-Hartley guinea pigs (200 g at the beginning of sensitization) were actively sensitized with OVA (grade III; Sigma) according to Andersson (1980): on two consecutive days, 20 μg of OVA together with 20 mg of AHG suspended in 0.5 ml of saline solution was administered i.p. to each animal.
Experimental Protocol and Drug Administration.
After 2 to 3 weeks, the animals were challenged with a single i.v. dose of 0.15 mg/kg OVA suspended in saline. The test compounds were either prepared as solutions or suspended in 4% methocel solution as described above (see Compounds), and administered i.v. by slow injection or p.o. by gavage, respectively, to the animals 1 h before OVA challenge. Control animals received the drug-free solvent i.v. or methocel solution p.o. OVA challenge and lung function measurements were performed in anesthetized (urethane, 1.2 g/kg i.p., 10 ml/kg, 30 min before challenge = −30 min) and mechanically ventilated animals. To abolish spontaneous breathing, the animals received pancuronium bromide (1 mg/kg i.v., −15 min). To enhance the allergen-mediated bronchoconstriction as described by Anderson et al. (1983), animals were pretreated with indomethacin (10 mg/kg i.v., −20 min), pyrilamine (2 mg/kg i.v., −6 min), and propranolol (0.1 mg/kg i.v., −5 min). Pyrilamine and propranolol were dissolved and diluted in saline, and indomethacin in 0.5 M NaHCO3 (10 mg/ml). The administration volume was 1 ml/kg i.v. and p.o. for all substances and drugs.
Measurement of the Allergen-Induced Bronchoconstriction.
The trachea of the anesthetized and cannulated animals was opened and a Y-shaped tracheal cannula (length 2.8 cm, outer diameter 3.1 or 2.5 mm) was inserted about 1.5 cm proximal to the bifurcation. About 15 min before OVA challenge, the animals received the muscle relaxant pancuronium bromide and were mechanically ventilated as described above (see Spasmogen-Induced Bronchoconstriction in the Anesthetized, Mechanically Ventilated Guinea Pig and BN Rat). The allergen-induced bronchoconstriction is characterized by a decrease of CON, which starts to change about 1 min after OVA injection and which reaches a plateau of 70 to 90% decrease after 4 to 6 min. All parameters were averaged at 1-min intervals from 30 to 1 min before OVA challenge and at 10-s intervals from 1 min before (prechallenge control phase) up to 12 min after OVA challenge (allergen-induced early, not histamine-mediated bronchoconstriction).
Data Analysis.
For biostatistical analysis, the delta-%-decrease of the values of the 1-min prechallenge control phase and the values at distinct 1-min time points from 1 to 12 min after challenge were calculated and the area under the curve (AUC) was determined for each animal. On basis of the mean AUC values, the inhibition of allergen-induced decrease of CON was calculated relative to placebo. After performing a multiple Student-Welch test to assess the significance of the differences between the groups, dose-response curves were calculated based on log-linear regression analysis to determine ED50 values; p < 0.05 was considered statistically significant.
Antigen-Induced Late Inflammatory Response in the BN Rat
Animals and Sensitization.
Male BN rats (180–220 g at the beginning of sensitization) were sensitized by the following method. Briefly, on day 1, 14, and 21, the rats received an s.c. (neck skin) injection of 0.5 ml of saline solution per rat containing 20 μg/ml OVA (grade V; Fluka) adsorbed to 40 mg/ml AHG; simultaneously, the animals were i.p. injected with 0.25 ml of B. pertussisvaccine per rat diluted in saline containing 4 × 108 heat-killed bacilli/ml.
Experimental Protocol and Drug Administration.
The drugs were administered p.o. as 4% methocel solution (seeCompounds) 18 h or 1 h before, 6 h or 24 h after OVA challenge. Nontreated OVA-challenged or nontreated nonchallenged controls received the equivalent amount of the corresponding solvent p.o. at 1 h before the OVA challenge (10 ml/kg of body weight for the drugs and placebo).
OVA challenge was performed 28 days after the beginning of sensitization. Each animal was placed in a special tube to restrain its activity and to guarantee nose-only exposure. These tubes were connected to an inhalation tower (CR equipment SA, Tannay, Switzerland), which allowed simultaneous challenge of 32 animals. The OVA-containing aerosol was generated by filtered air at a pressure of 1.7 bar (600 l/h) using a medication nebulizer device (Hospitak; delivered by Carbamed, Basel, Switzerland) and a solution of 3.2 mg/ml OVA (grade V; Sigma; diluted in saline). The aerosol was moved forward by a continuous air flow of 2000 l/h. The exposure time was 1 h, resulting in an aerosolized OVA volume of about 20 ml. The nonchallenged controls (sham challenge) were sensitized with OVA and exposed to saline aerosol. Forty-eight hours after OVA/sham challenge, animals were anesthetized with Trapanal (thiopental, 150 mg/ml; diluted in Aqua dest.; 1 ml/animal) and BAL was performed. The trachea of the rats was then exposed and cannulated, followed by gently lavaging the lungs three times in situ with 4 ml/animal (∼2 ml/100 g of body weight) BAL buffer (phosphate-buffered saline solution containing 0.372% sodium EDTA). On average, 80% of the administered BAL fluid was recovered regardless of pretreatment.
Cell Differentiation, and Determination of Total Protein and TNFα in BALF.
Total cell counts and cell type differentials in BALF of placebo-treated/OVA-challenged and drug-treated/OVA-challenged rats were obtained immediately after BAL using an automatic leukocyte differentiation system (Cobas Helios 5 Diff; Hoffmann-La Roche AG). For this purpose, BAL samples (about 3 ml) were directly applied to the analyzer and were automatically mixed with Eosinofix that lyses red blood cells, stabilizes membranes of leukocytes, and stains eosinophils. This procedure allows the two-dimensional differentiation of leukocytes by size (measured by a change in resistance between two electrodes) and optical density (measured by scatter light). The adaptation and validation of this automatic system for the measurement of rat BALF from OVA-challenged animals was described previously (Hatzelmann et al., 1996).
For nonchallenged controls (sham challenge), total cell counts were determined using the cell counter Sysmex F-300 (Digitana AG, Hamburg, Germany) adjusted to count white blood cells. In parallel, leukocyte differentiation was done on Diff-Quick-stained smears prepared by cytocentrifuging 100 μl of BALF containing infiltrated cells (∼300 cells/μl) at 190 g for 2 min (Cytospin 2; Shandon Inc., Pittsburgh, PA).
Protein concentration in cell-free BALF was measured using a commercially available protein assay (Dye Reagent Concentrate; Bio-Rad Laboratories GmbH, München, Germany). For standardization, bovine serum albumin was used.
Determination of TNFα in cell-free BALF was performed by ELISA (QuantikineM, Rat TNFα immunoassay; R&D Systems, Minneapolis, MN; detection limit: 12.5 pg/ml). Previous internal studies showed peak TNFα concentration in BALF at 48 h after OVA-challenge (D. S. Bundschuh, unpublished data).
Data Analysis.
The experiment consisted of several drug-treated groups and two solvent-treated control groups (OVA-sensitized/sham-challenged, OVA-sensitized/OVA-challenged), at least eight animals per group. The drug-induced individualchanges were calculated comparing the drug-treated with the solvent-treated OVA-challenged control animals [effect = {100% × (drug-treated OVA-challenged individual value) − (median OVA-challenged control)}/{(median OVA-challenged control) − (median sham-challenged control)} = 100% × Δdrug/Δcon]. Biostatistical analysis of the data was performed based on medians. The Mann-Whitney test (a modified Wilcoxon Rank Sum test) was used to determine significant differences with regard to the OVA-challenged control for each experiment. The delta %-change of Δdrug/Δcon (see above) represents the individual relative, drug-induced change with regard to the change of the controls. The half-maximum effective dose (ED50) was taken from fitted dose-response curves by log-linear regression analysis based on the individual inhibition values using the above-mentioned ratios with limits of 0 and 100% inhibition (GraphPad Prism 2.01; GraphPad Software, Inc., San Diego, CA). Monotone dose dependence was evaluated by the Jonckheere Terpstra test using all doses tested. In the case of significance (p < 0.05), the highest dose was considered to show a statistically significant increased inhibition in comparison to the lowest dose. Subsequently, the Jonckheere Terpstra test was repeated without the highest dose and the evaluation was performed again as described above; p < 0.05 was considered statistically significant.
LPS-Induced TNFα Release in the Sprague-Dawley Rat
Animals and Drug Administration.
The experiments were performed using male Sprague-Dawley rats (200–250 g). Test compounds were suspended in 4% methocel as described above (seeCompounds) and given p.o. by gavage (10 ml/kg of body weight). Control animals were treated with the drug-free methocel solution.
LPS-Challenge and TNFα Determination.
One hour after compound administration, LPS (S. abortus equi; diluted in saline containing 0.1% hydroxylamine) was i.v. injected at the dose of 0.1 mg/kg (1 ml/kg). Final anesthesia was performed i.v. with a pentobarbital solution (48 mg/kg) containing heparin (1000 IU/kg) 1.5 h after LPS challenge. Heparinized blood was obtained by heart puncture. Blood was centrifuged and plasma samples were kept frozen at −80°C until determination of TNFα levels by ELISA (QuantikineM, Rat TNFα immunoassay, R&D Systems; detection limit: 12.5 pg/ml).
Data Analysis.
Suppression of TNFα production was calculated individually in percentage for each drug-treated animal in relation to the median TNFα level of the vehicle-treated LPS-challenged control group of the same experiment. The dose-response curves based on the mean values of 8 to 16 animals/dose were calculated using the nonlinear regression analysis provided with the GraphPad Prism software package (GraphPad Software, Inc.) with limits of 0 and 100% inhibition. ED50 (effective dose with 50% inhibition) values and 95% confidence limits were derived from these dose-response curves; p < 0.05 was considered statistically significant.
Results
Inhibition of Antigen-Induced Trachea Contraction by Roflumilast.
This ex vivo study was performed to assess the potency of roflumilast to inhibit antigen-induced contractions of isolated tracheal chains prepared from OVA-sensitized guinea pigs known to be sensitive to selective PDE4 inhibitors (Underwood et al., 1993,1998). In tracheal chains, cumulative administration of OVA in one-log unit steps from 10−10 to 10−7 g/ml produced concentration-dependent contractions of maximally 70 to 80% of those elicited by 10−3 M carbachol (Fig.1). During antigen-evoked trachea contraction, two more or less separable phases of contraction could be observed: a transient peak component (within 0–3 min), which has previously been correlated with histamine release (preformed mediator), and a more stable and tonic component (within 8–15 min), which is known to be closely associated with eicosanoid synthesis and release (Undem et al., 1988; Underwood et al., 1993).
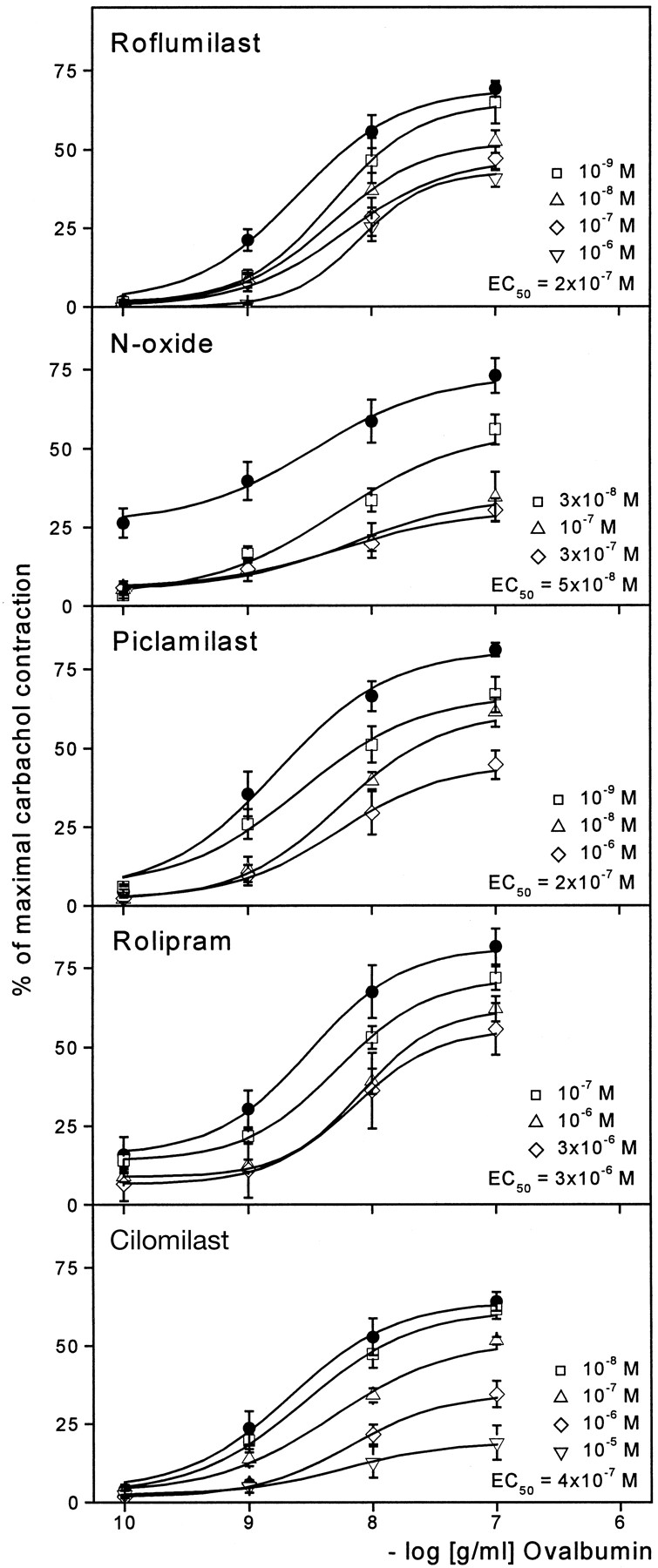
Effect of increasing concentrations of roflumilast, roflumilast N-oxide, piclamilast, rolipram, and cilomilast (from top to bottom) on OVA-evoked contractions of isolated tracheal chains prepared from OVA-sensitized guinea pigs. Filled circles (●) mark the maximal antigen-induced contraction. All contraction values are expressed as percentage of reference contraction induced by 10−3 M carbachol. Results are given as means ± S.E.M.; n = 6–10 tracheal chains in each case. Displayed are approximate EC50 values for half-maximal inhibition calculated by linear regression.
A 30-min pretreatment of the trachea with roflumilast (10−9–10−6 M) caused a concentration-dependent inhibition of the antigen-induced tonic contraction (Fig. 1). Due to a concomitant rightward shift of the OVA concentration-response curves, the effect of lower OVA concentrations was more attenuated by roflumilast than that of higher antigen concentrations. A concentration of 2 × 10−7 M of roflumilast was necessary to inhibit submaximal contractions evoked by 10−8 g/ml OVA by approximately 50%. Similarly, the N-oxide metabolite of roflumilast at 3 × 10−8 and 10−7 M inhibited antigen-evoked contractions in a concentration-dependent manner with an EC50 of 5 × 10−8 M, an inhibitory effect that, however, could not be further enhanced by higher concentration (3 × 10−7 M). Also piclamilast (10−9–10−6 M) and cilomilast (10−8–10−5 M) antagonized OVA-induced trachea contractions with EC50 values of 2 × 10−7 M and 4 × 10−7M, respectively, whereas rolipram (10−7–3 × 10−6 M) proved to be approximately 10-fold less potent (EC50 = 3 × 10−6 M) (Fig. 1). Roflumilast and itsN-oxide (up to 10−6 M) caused no or only weak relaxation, respectively, of guinea pig tracheal chains contracted spontaneously (data not shown).
Inhibition of Spasmogen-Induced Bronchoconstriction by Roflumilast.
The aim of these investigations was to determine the direct effect of roflumilast on spasmogen-mediated smooth muscle contraction in the small and large airways of anesthetized, mechanically ventilated BN rats and guinea pigs. Intravenously administered roflumilast dose and time dependently attenuated the serotonin- or histamine-induced decrease of airway conductance in BN rats (Fig. 2) or guinea pigs (Fig.3), respectively. As depicted, the bronchodilatory activity of roflumilast was most pronounced at 2 min after compound administration. ED50 values display the inhibition of bronchoconstriction by roflumilast at 60 min post-treatment. For comparison, the bronchospasmolytic activity of cilomilast in the BN rat model proved to be 2 times weaker than roflumilast (ED50 = 10.8 μmol/kg i.v.; individual data not shown).
Inhibition of Antigen-Induced Bronchoconstriction by Roflumilast.
Additional experiments were conducted to assess the effect of roflumilast on early, not histamine-mediated bronchoconstriction evoked by antigen. In anesthetized guinea pigs, antigen injected i.v. caused an acute bronchospasm, which peaked within 4 to 6 min (70–90% decrease of conductance). Pretreatment of the animals with i.v. administered roflumilast inhibited this bronchoconstriction in a time- (data not shown) and dose-dependent manner (Table 1). Oral bioavailability of roflumilast was demonstrated with a 14 times lower potency compared with the i.v. route (Table 1). The orally given N-oxide of roflumilast, the primary metabolite, was equally effective in this model and therefore is likely to contribute to the antiallergic activity of roflumilast in vivo. Compared with the reference PDE4 inhibitors, roflumilast showed a 6-, 22-, and 35-fold superiority to piclamilast, rolipram, and cilomilast, respectively. The corresponding ED50 values are listed in Table2.
Inhibitory effects of i.v. and p.o. roflumilast on allergen-induced early airway reaction in the guinea pig
Inhibitory effects of roflumilast and reference PDE4 inhibitors on allergen-induced early airway reaction in the guinea pig
Inhibition of Antigen-Induced Late Inflammatory Airway Response by Roflumilast
Inhibition of Cell Infiltration and Protein Accumulation by Roflumilast.
To evaluate the anti-inflammatory potential of orally administered roflumilast, OVA-induced cell infiltration and total protein concentration in BALF of OVA-sensitized BN rats were determined. In averaging all saline- and antigen-challenged control groups used in these pharmacological studies, total cell counts increased from 376 to 460 total cells/μl (consisting of 2–16% eosinophils, 1–7% neutrophils, 13–19% lymphocytes, 59–82% macrophages) to 1571 to 3200 cells/μl at 48 h postchallenge. This 4- to 7-fold increase in total cell numbers was based on a 12- to 189-fold increase in eosinophils, an 11- to 118-fold increase in neutrophils, a 6- to 10-fold increase in lymphocytes, and a 1.4- to 1.7-fold increase in macrophages, accounting for 46 to 47% eosinophils, 19 to 21% neutrophils, 16 to 22% lymphocytes, and 19 to 20% macrophages. Additionally, the protein concentration of the OVA-challenged animals was about 8 to 12 times higher than that of nonchallenged controls (1.54–2.61 and 0.13–0.26 mg/ml protein, respectively). Roflumilast completely inhibited eosinophil (Fig.4A) and neutrophil (Fig. 4B) infiltration as well as microvascular leakage (Fig. 4C) at an oral dose of 10 μmol/kg; significant inhibition (61–80%; p < 0.01) of these parameters was still observed at 3 μmol/kg roflumilast. Roflumilast N-oxide was equally effective as its mother compound, whereas piclamilast and rolipram showed a 6- to 9-fold lower potency than roflumilast in inhibiting cellular and protein influx (Fig. 4). Displaying an even bigger difference, cilomilast was about 1.5 orders of magnitude weaker in potency compared with roflumilast (Fig. 4). For a quantitative analysis of the PDE4 inhibitors tested, ED50 values for all parameters measured were calculated and summarized in Table 3.

Inhibition of antigen-induced eosinophil (A) and neutrophil (B) influx into alveolar space and protein (C) accumulation in BALF of OVA-challenged BN rats by p.o. administered roflumilast and reference PDE4 inhibitors. Results are given as means ± S.E.M.;n = 16–32 (roflumilast), n = 24–32 (N-oxide), n = 8–24 (piclamilast), n = 8 (rolipram),n = 16–32 (cilomilast) animals/compound dose,n = 12/placebo group; 3–7 doses/compound.
Inhibitory effects of roflumilast and reference PDE4 inhibitors on late airway inflammation in the BN rat
Inhibition of TNFα Release by Roflumilast.
Pharmacological evidence is given for the fact that the proinflammatory cytokine TNFα mediates the recruitment of neutrophils and eosinophils during airway inflammation in mice and rats (Lukacs et al., 1995; Renzetti et al., 1996). To test whether TNFα production in a model of allergic lung inflammation is affected by roflumilast, we determined TNFα concentrations in the BALF of BN rats 48 h after OVA challenge. Typically, in OVA-sensitized rats, challenge resulted in a liberation of 225.4 ± 28.2 pg/ml TNFα (mean ± S.E.M.;n = 8; one representative of 20 independent experiments), whereas TNFα concentrations were below the detection limit in BALF of OVA-sensitized but nonchallenged animals in all experiments (data not shown). Orally administered roflumilast dose dependently decreased the release of TNFα into the alveolar lumen of challenged BN rats with an ED50 of 1.2 μmol/kg and was similar to its N-oxide (ED50 = 1.6 μmol/kg) but 6- to 59-fold more effective than the reference PDE4 inhibitors as depicted in Fig.5 and Table 3.

Inhibition of TNFα release into alveolar space (BALF) of OVA-challenged BN rats by p.o. administered roflumilast and reference PDE4 inhibitors. Results are given as means ± S.E.M.;n = 8–16 animals/compound dose; 3–5 doses/compound.
Roflumilast Time Course Study.
We next carefully addressed the question as to what extent pre- and post-treatment with roflumilast is effective in our model of late airway inflammation. Therefore, we compared different time points of roflumilast administration, i.e., 18 and 1 h before, and 6 and 24 h after challenge. As illustrated in Fig. 6, A and B, the effect of 1-h pretreatment of OVA-sensitized/challenged animals with roflumilast (10 μmol/kg p.o.) on cell infiltration into alveolar space was most pronounced (suppression of eosinophil and neutrophil influx by 91 and 98%, respectively). Since the 18-h pretreatment still resulted in a 68 and 81% inhibition of eosinophil and neutrophil influx, respectively, the duration of action of orally administered roflumilast was more than 18 h. Looking at post-treatment regimens, antigen-induced cellular response was significantly affected by roflumilast even when given 6 h after challenge. Interfering as late as 24 h postchallenge, an inhibitory effect of roflumilast on neutrophil but not eosinophil influx was observed. Intriguingly, the release of TNFα into the alveolar lumen was completely inhibited at all administration time points investigated (Fig. 6C).

Inhibition of eosinophil (A) and neutrophil (B) influx, and TNFα (C) release into alveolar space of OVA-challenged BN rats by roflumilast (10 μmol/kg), p.o. administered 18 or 1 h before (−18 h, −1 h), and 6 or 24 h after (+6 h, +24 h) OVA challenge. The graphs display absolute values determined in sensitized/nonchallenged/nontreated (OVA/−/−), sensitized/challenged/nontreated (OVA/OVA/−), and sensitized/challenged/roflumilast-treated (OVA/OVA/roflumilast) animals 48 h after challenge. Statistical analyses were performed using the Mann-Whitney test comparing each group with the corresponding OVA-challenged control as described under Experimental Procedures. Results are given as means ± S.E.M.;n = 8 animals/treatment group,n = 12 for placebo. **p < 0.01, ***p < 0.001 versus OVA/OVA/−;###p < 0.001 versus OVA/−/−.
Inhibition of LPS-Induced TNFα Release by Roflumilast
Finally, the influence of roflumilast, its N-oxide, and the reference PDE4 inhibitors on LPS-induced TNFα release, a well characterized cAMP-sensitive pathway in cells of the monocytic/macrophage lineage, was investigated in vivo. Roflumilast as well as its N-oxide metabolite suppressed the production of TNFα in the rat with an ED50 of 0.3 μmol/kg, whereas piclamilast, rolipram, and cilomilast were about 9, 25, and 310 times less potent (Table 4).
Suppression by roflumilast and reference PDE4 inhibitors of LPS-induced circulating TNFα in the rat
Discussion
The objective of this study was to establish a comprehensive profile of the in vivo airway pharmacology of roflumilast, a novel, orally active PDE4 inhibitor. Therefore, we evaluated its bronchodilatory, anti-inflammatory, and antiallergic potential in several disease models in guinea pigs and rats in direct comparison with other PDE4-specific compounds. As reference drugs we chose piclamilast (RP 73401) (Ashton et al., 1994) for its structural proximity to roflumilast, rolipram as the archetype PDE4 inhibitor (Underwood et al., 1993), and cilomilast as the most advanced drug of its class (Barnette, 1999). Since roflumilast N-oxide was found to be the primary metabolite in several animal species and humans (M. David, E. Sturm, and K. Zech, manuscript in preparation), it was of interest to investigate whether this metabolite is active by itself and therefore may contribute to the pharmacological effects in vivo of roflumilast.
Roflumilast possesses a broad range of pharmacological activities of potential use in the treatment of airway disorders. It not only potently modulates the activity of various inflammatory cells in vitro (cf. Hatzelmann and Schudt, 2001) but also blocks inflammatory and pathophysiological cascades in complex in vivo settings. The data presented here demonstrate the bronchodilator activity of roflumilast in isolated tracheal strips ex vivo, and in allergic guinea pigs and rats in vivo, and its ability to suppress the generation of a key proinflammatory cytokine, TNFα, in rats. In addition, roflumilast very impressively inhibits pulmonary eosinophilia as well as microvascular leakage, supporting the anti-inflammatory potential of roflumilast. Overall, roflumilast appears to be the most potent PDE4-selective inhibitor tested, even in comparison with the most advanced second-generation inhibitor cilomilast presently undergoing clinical trials for asthma and COPD.
We demonstrated that roflumilast, its N-oxide metabolite, and the reference molecules all potently attenuated antigen-evoked contractions of tracheal chains, whereas these drugs exerted negligible effects on trachea contractions occurring spontaneously (this study) or by exogenous spasmogens (Underwood et al., 1993, 1998). Thus, the increased potency of roflumilast in allergen-driven pathological situations might be attributed to inhibition of mast cell degranulation rather than to direct smooth muscle relaxation (Underwood et al., 1993). In agreement with that notion, the bronchospasmolytic effect of roflumilast in vivo was less pronounced compared with the suppression of the allergen-induced early, not histamine-mediated bronchoconstriction (Figs. 2 and 3; Table 1).
In vitro, roflumilast selectively inhibits PDE4 activity and activation of various leukocytes involved in inflammation (cf. Hatzelmann and Schudt, 2001). Thus, it was of interest to find out whether orally administered roflumilast exerts anti-inflammatory effects in vivo. Infiltration and accumulation of inflammatory cells as well as edema formation, commonly seen features associated with human bronchial asthma and also to be observed in OVA-sensitized and OVA-challenged BN rats (Elwood et al., 1991; Raeburn et al., 1994; Schneider et al., 1997), were effectively inhibited by roflumilast, itsN-oxide metabolite, and all reference PDE4 inhibitors tested (Table 3). Whereas eosinophils play a pivotal role in the pathophysiology of asthma, COPD is marked by an increase in the activation and number of neutrophils. Lymphocytes, especially T cells, are crucial in initiating and orchestrating ongoing immunologically driven chronic asthma. The latter cells as well as eosinophils themselves are potential sources of cytokines that may influence eosinophil function, whereas activation of neutrophils results in the release of numerous mediators and proteases that contribute to the progression of inflammation, fibrosis, and tissue destruction (Corrigan and Kay, 1992). It is evident that all inflammatory parameters investigated were inhibited by all PDE4 inhibitors at the same relative potency (Table 3). However, roflumilast and its N-oxide metabolite displayed by far (about 6- to 40-fold) the highest potency. The discrepancy within the benzamides (roflumilast, N-oxide, piclamilast), i.e., similar potency in vitro (cf. Hatzelmann and Schudt, 2001) but superior potency of roflumilast and itsN-oxide metabolite in vivo, is probably due to a weak oral bioavailability of piclamilast, one of the reasons why this compound was mainly investigated intratracheally and i.v. in vivo (Raeburn et al., 1994) and was in clinical development as an inhalative agent for the treatment of asthma (Jonker et al., 1996). In addition, in both antigen-induced animal models investigated, cilomilast turned out to be the PDE4 inhibitor with the lowest potency. This is in line with the in vitro results (cf. Hatzelmann and Schudt, 2001) showing that IC50 values for the inhibition of leukocyte cell functions were consistently higher for cilomilast compared with benzamide PDE4 inhibitors. The reason for this different effectiveness 1) is the higher potency of benzamides with regard to inhibition of PDE4 activity; and 2) might be due to differences in the discrimination between low-affinity rolipram binding site and high-affinity rolipram binding site, an issue further discussed in the accompanying paper byHatzelmann and Schudt (2001).
The efficacy of roflumilast post-treatment in antagonizing eosinophilia and inhibiting TNFα synthesis induced by antigen-challenge supports the suggestion that roflumilast is affecting eosinophil and neutrophil trafficking and/or activation rather than inhibiting mast cell degranulation. By increasing intracellular cAMP levels, activated cells may be inhibited from discharging their contents (mediator and protease release). This hypothesis is confirmed by the in vitro effects described for roflumilast (cf. Hatzelmann and Schudt, 2001) and is in agreement with the results found for the reference PDE4 inhibitor cilomilast (Underwood et al., 1998).
The proinflammatory cytokine TNFα plays an important role in the initial phase of the inflammatory response. It was found in BALF and sputum of allergic asthmatics (Virchow et al., 1995; Keatings et al., 1996), which suggests that it may play a role in the late-phase airway response and cell recruitment. Moreover, pharmacological evidence is given for the fact that TNFα mediates the recruitment of neutrophils and eosinophils during antigen-induced airway inflammation (Lukacs et al., 1995; Renzetti et al., 1996). However, the inhibition of eosinophil recruitment by TNFα receptor fusion protein was less pronounced compared with neutrophils (Renzetti et al., 1996). In agreement with this report, our time course studies showed that roflumilast treatment 24 h postchallenge completely inhibited TNFα release and in parallel significantly reduced neutrophil accumulation but failed to influence the eosinophil influx (Fig. 6), suggesting additional factors besides TNFα contributing to the control of late eosinophil recruitment into the lung during allergic inflammation.
Since COPD is characterized by an increase in the activation and/or number of not only neutrophils and CD8+ T cells but also macrophages (Barnes, 2000), it was important to show the efficacy of roflumilast in a macrophage-dependent in vivo model. Moreover, concentrations of TNFα were reported to be increased in the sputum of patients with COPD (Keatings et al., 1996). Indeed, roflumilast, its N-oxide metabolite and the reference PDE4 inhibitors proved to be highly active against the generation of LPS-induced TNFα, a cAMP-sensitive pathway highly attenuable by PDE4 inhibitors (Fischer et al., 1993). The rank order of potency found for this anti-inflammatory effect was roflumilast =N-oxide > piclamilast > rolipram > cilomilast, mirroring the effectiveness observed in the allergen-induced airway disease models in the guinea pig and the BN rat. The apparent discrepancy of p.o. ED50 values found with cilomilast in this study (ED50 = 93 μmol/kg ≡ 31.9 mg/kg) compared with data previously shown byGriswold et al. (1997; ED50 = 0.4 mg/kg) might be due to differences in the experimental design (e.g., SD rats versus Lewis, LPS from S. abortus equi versus Escherichia coli, LPS injection i.v. versus i.p., drug administration 60 versus 30 min before LPS-challenge) and/or the ELISAs used to detect rat TNFα (anti-ratTNFα antibodies versus anti-murineTNFα antibodies).
The therapeutic potential of TNFα inhibition in a variety of chronic inflammatory diseases is widely accepted, and various strategies for TNFα inhibition have been suggested (Newton and Decicco, 1999). Furthermore, a TNFα-modulating/controlling potential is attributed to PDE4 inhibitors (Gantner et al., 1997). In agreement with these findings, our results provide further evidence that roflumilast exhibits a strong anti-inflammatory potential to be of therapeutic use not only for chronic airway diseases but also for other inflammatory disorders.
In addition, roflumilast has other favorable characteristics. Time course studies presented here demonstrate that roflumilast significantly inhibited allergen-induced cell infiltration and TNFα release for at least 18 h after administration. These data indicate that roflumilast possesses a prolonged duration of action comparable with cilomilast (Griswold et al., 1998). Moreover, multiple dosing of roflumilast for 4 days b.i.d. did not reveal diminution of activity (data not shown). Therefore, as described for cilomilast, but in contrast to rolipram (Griswold et al., 1998), no evidence for induction of pharmacological tolerance can be attributed to roflumilast.
In summary, the preclinical data presented in this study indicate a spectrum of potential activities for roflumilast in the treatment of asthma and COPD. Since the in vivo efficacy of roflumilast investigated also translates to its main metabolite (N-oxide) in several mammalian species, it is likely that the N-oxide also contributes to the overall pharmacological action of roflumilast in humans. This combination of bronchodilatory, antiallergic, and anti-inflammatory properties is unique and unparalleled by other classes of drugs in use for chronic respiratory disorders. Roflumilast is currently under investigation in clinical trials covering COPD (phase II) and asthma (phase III) to prove beneficial efficacy in patients. These clinical studies will reveal whether roflumilast fulfills its therapeutic promise.
Acknowledgments
We thank Dr. U. Kilian, who established the respiratory pharmacology group at Byk Gulden. We also thank Dr. L. Mazzoni (Novartis, Basel, Switzerland) for advisory and methodological support in the set up of the BN rat model and Dr. F. Gantner for fruitful discussions. The excellent technical assistance of G. Dillmann, U. Graf, H. König, S. Kuklinski, C. Orjeda, A. Ostermann, M. Stade, M. Uhr, and G. Wardsack is highly appreciated.
Footnotes
-
Send reprint requests to: Dr. Daniela S. Bundschuh, Department of Pharmacology, Byk Gulden, P.O. Box 10 03 10, 78403 Konstanz, Germany. E-mail: daniela.bundschuh{at}byk.de
-
This work is dedicated to the inventor of roflumilast, Dr. Hermann Amschler, who deceased in 1999 to our deepest regret.
- Abbreviations:
- COPD
- chronic obstructive pulmonary disease
- PDE4
- phosphodiesterase type 4
- roflumilast
- 3-cyclopropylmethoxy-4-difluoromethoxy-N-[3,5-dichloropyrid-4-yl]-benzamide
- cilomilast
- Ariflo, SB 207499
- N-oxide
- roflumilastN-oxide
- piclamilast
- RP 73401
- BN
- Brown Norway
- OVA
- ovalbumin
- LPS
- lipopolysaccharide, endotoxin
- PEG400
- polyethylene glycol 400
- AHG
- Al(OH)3
- F
- flow
- PIP
- inflation pressure
- TV
- tidal volume
- RAW
- airway resistance
- CON
- conductance
- AUC
- area under the curve
- BAL
- bronchoalveolar lavage
- TNFα
- tumor necrosis factor-α
- BALF
- bronchoalveolar lavage fluid
- ELISA
- enzyme-linked immunosorbent assay
- Received September 15, 2000.
- Accepted December 4, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}