


Abstract
The ability of the second generation phosphodiesterase 4 inhibitor SB 207499 (Ariflo), [c-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)-r-l-cyclohexane carboxylic acid], to inhibit inflammatory cytokine production in vivo was evaluated and compared to that of rolipram, a first generation phosphodiesterase 4 inhibitor. To examine human tumor necrosis factor alpha (TNFα) production, human monocytes were adoptively transferred into Balb/c mice and challenged with lipopolysaccharide (LPS). In this model, SB 207499 inhibited human TNFα production with oral ED50 of 4.9 mg/kg. Similarly, R-rolipram inhibited human TNFα production with an ED50of 5.1 mg/kg, p.o. In contrast to their equipotent activity against TNFα production, SB 207499 (ED50 = 2.3 mg/kg, p.o.) was 10-fold less potent than R-rolipram (ED50 = 0.23 mg/kg, p.o.) in reversing reserpine-induced hypothermia, a model of antidepressant activity. In time course studies, SB 207499 (30 mg/kg, p.o.) inhibited TNFα production for at least 10 hr; substantial plasma concentrations of SB 207499 were detected over the same interval. The ability of SB 207499 to modulate interleukin-4 productionin vivo was assessed in a chronic oxazolone-induced contact sensitivity model in Balb/c mice. In this model, topical administration of SB 207499 (1000 μg) inhibited intralesional concentrations of interleukin-4 (55%; P < .01). The results demonstrate that SB 207499 is a potent inhibitor of inflammatory cytokine production in a variety of settings in vivo. Moreover, although it is as potent as R-rolipram in inhibiting TNFα production, it has substantially less central nervous system activity. Thus SB 207499 represents an excellent candidate with which to evaluate the antiinflammatory potential of PDE4 inhibitors.
The role of cyclic AMP as a second messenger involved in the suppression of immune and inflammatory cell activity is well-documented (Bourneet al., 1974, Plaut et al., 1980; Torphy, 1998). Although agents that increase cyclic AMP content inhibit the generation or release of a host of inflammatory mediators, considerable attention has focused on the regulation of inflammatory cytokines, particularly TNFα. This cytokine is implicated in the pathogenesis of a number of inflammatory disorders, including asthma (Anticevich et al., 1995), rheumatoid arthritis (Feldmann et al., 1995), multiple sclerosis (Navikas et al., 1996) and endotoxic shock (Bellomo, 1992). The elaboration of TNFα from monocytes is strongly inhibited by prostacyclin analogs or PDE inhibitors (Giembycz and Dent 1993; Eisenhut et al., 1993; Prabhakar et al., 1994), agents that increase cyclic AMP content.
The low Km cyclic AMP-specific phosphodiesterase (PDE4), one of seven genetically distinct families of PDEs, is the major cyclic AMP-metabolizing enzyme in nearly all immune and inflammatory cells (Torphy and Undem, 1991; Giembyez and Dent, 1993). This isozyme family is selectively inhibited by a number of compounds, including rolipram (Davis, 1984), denbufylline (Nicholson et al., 1989), tibenelast (Ho et al., 1990) and CP 80633 (Cohan et al., 1996). Predictably, these prototypical first generation isozyme-selective PDE4 inhibitors produce a broad spectrum of anti-inflammatory effects in both in vitro and in vivo settings (Griswold et al., 1993; Torphy and Undem, 1991; Giembycz and Dent, 1993; Torphy, 1998). With respect to TNFα, a number of PDE4 inhibitors suppress the production of this cytokine from LPS-stimulated peripheral blood monocytes or peritoneal macrophages (Semmler et al., 1993; Prabhakar et al., 1994;Seldon et al., 1995; Barnette et al., 1996). Although information on the anticytokine activity of PDE4 inhibitorsin vivo is less plentiful, two first generation PDE4 inhibitors, rolipram and BRL 61063, have been shown to reduce endogenous or stimulus-generated TNFα production in mice and rats (Badger et al., 1994; Kaplan et al., 1995; Turneret al., 1993). Collectively, this information has raised hopes that PDE4 inhibitors will be useful in treating a number of acute and chronic inflammatory diseases.
Despite the attractive antiinflammatory profile of PDE4 inhibitors, the therapeutic activity of first generation agents is limited by their side effects. The most prominent of these are nausea and vomiting (Horowski and Sastre-Y-Hernandez, 1985). These side effects are largely attributable to CNS actions of PDE4 inhibitors, although local activity on the gastrointestinal tract may also be involved (Heaslip and Evans, 1995). Regarding the latter point, PDE4 inhibitors are particularly potent stimulators of acid secretion from gastric parietal cells (Barnette et al., 1995).
SB 207499 (Ariflo) [c-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)-r-l-cyclohexanecarboxylic acid] is among the first of a new generation of highly selective and potent PDE4 inhibitors specifically designed to have an improved therapeutic index compared with first generation compounds (Christensenet al., 1998; Torphy et al., 1997; Barnetteet al., 1998). SB 207499 is currently being evaluated in clinical trials for the treatment of asthma. The molecular basis for the improved therapeutic index is related in part to its decreased potency against a unique conformer of PDE4 that appears to be enriched in the CNS and parietal cells (Torphy et al., 1992, Jacobitzet al., 1996; Barnette et al., 1997; Souness and Rao, 1997). Indeed, previous studies indicate that SB 207499 is equipotent to R-rolipram as an inhibitor of LPS-induced TNFα production from isolated human monocytes, but is at least 100-fold less potent as an inhibitor of H+ secretion from isolated parietal cells (Barnette et al., 1998).
Against this backdrop, we profiled the in vivo activity of SB 207499 in the mouse with the following objectives: 1) evaluate the oral activity of SB 207499 against LPS-stimulated TNFα in a human monocyte adoptive transfer model and a model of endotoxic shock, 2) evaluate the relationship between pharmacodynamics and plasma concentrations of SB 207499, 3) demonstrate the ability of SB 207499 to retain its activity after repeat dosing, 4) evaluate the ability of SB 207499 to inhibit IL-4 production in vivo and 5) compare the therapeutic ratio of SB 207499 with that of the first generation PDE4 inhibitor, rolipram. The results indicate that SB 207499 is a potent, long-lasting and orally active inhibitor of inflammatory cytokine production in vivo.
Methods
Animals.
Balb/c, CD-1 and C57B1/6 male mice were obtained from Charles River Breeding Laboratories, Wilmington, MA and The Jackson Laboratories, Bar Harbor, MA, respectively and were maintained in a barrier-sustained facility. Age-matched animals were used in the weight range from 18 to 25 g. Groups of from three to nine animals were used for these studies. All protocols were approved by the Animal Care and Use committee, SmithKline Beecham Pharmaceuticals (King of Prussia, PA).
Isolation of human monocytes.
Monocytes were isolated from fresh human whole blood obtained via venepuncture or Source Leukocyte packs (Biological Specialties, Inc., Lansdale, PA) using the following procedure performed at 25°C. Polymorphonuclear leukocytes were separated by layering the blood on Histopaque-1077 (Sigma Chemical Co., St. Louis, MO) with centrifugation at 800 × g for 30 min. The lymphocyte/monocyte portion was harvested and washed twice with DPBS at 250 × g for 10 min. The pellet was resuspended in 13 ml DPBS, layered on 13 ml Percoll solution (Sigma) prepared in RPMI-1640 media (Gibco, Grand Island, NY), and centrifuged at 550 × g for 30 min. The buoyant layer of monocytes was removed and washed twice with DPBS. The monocyte preparations normally were 70% monocytes (ranged from 65 to 90%) and the viabilities were >97% by trypan blue exclusion.
In vivo administration of human monocytes for the production of TNFα.
The method used to assess simultaneously the inhibitory actions of compounds on human and mouse TNFα production was previously described (Griswold et al., 1996). Briefly, Balb/C male mice in groups of three to eight were injected with 0.25 ml or 0.5 ml of either saline, DPBS or 2 to 10 × 106human monocytes per ml DPBS (depending on the experimental need and/or preparation) into the peritoneum using light pressure on a syringe with a 23-gauge needle so that the monocytes are exposed to minimal shearing forces and stress. Two min after receiving monocytes, mice used in pharmacological studies were treated with compound (10 ml/kg dose volume) or the vehicle. Fifteen or 30 min later the animals received injections i.p. with 25 μg LPS (0.2 ml of 125 μg endotoxin per ml DPBS; Escherichia coli, type W, Difco-Laboratories, Detroit, MI), or an amount of LPS specified. Two hours later, the animals were euthanized by carbon dioxide asphyxiation and 1.5 ml DBPS (4°C) was inoculated, i.p. The peritoneum was gently massaged and the wash was removed and placed in polypropylene microtubes in an ice bath. The samples were clarified by centrifugation at 12,500 × gfor 5 min, 4°C. The supernatants were decanted and assayed for human and mouse TNFα by ELISA developed at SmithKline Beecham (Oliveraet al., 1992). The sensitivity for the ELISA is 23 to 1000 pg/ml for human TNFα and 25 to 800 pg/ml for mouse TNFα.
Duration of action studies.
Evaluation of the duration of action was accomplished using LPS-induced TNFα production in Balb/c mice as described above. Animals (three to six/group) were given SB 207499 at different pretreatment times and plasma samples were obtained for assay of TNFα levels as described.
Reversal of reserpine-induced hypothermia.
Reversal of reserpine-induced hypothermia is a standard model used to assess the psychotropic activity of compounds (Sanghvi and Gershon, 1977). Male Balb/c mice (20–30 g) were individually isolated in wire cages. The rectal temperature of each mouse was recorded before pretreatment with reserpine (10 mg/kg, i.p.). Four hours after reserpine the rectal temperatures were recorded again and individual animals were given by gavage vehicle (25% polyethylene glycol, PEG; 0.1 ml/10 mg body weight) or various doses of SB 207499 dissolved in 25% PEG. Rectal temperatures were then recorded every 30 min for 2 hr. Dose-response curves were constructed using the data obtained 90 min after drug treatment, and the data expressed as the increase in temperature from that observed at 4 hr after reserpine (temperatures were reduced by approximately 15.4 ± 0.14°C below basal temperature). ED50 values were determined by probit analysis of the data obtained from the mean values of eight or nine animals. Racemic rolipram was used as an internal standard.
Pharmacokinetic studies.
SB 207499 was prepared at 3 mg/ml in extra virgin olive oil on the day of dosing. Three male mice per time point were dosed by gavage (10 ml/kg). After they were killed by carbon dioxide asphyxiation, blood samples (0.4–1.0 ml) were collected at 0.25, 0.5, 1, 2.5, 4, 6, 8 and 10 hr postadministration. The blood samples were collected into labeled, heparinized tubes and kept on ice. Plasma was obtained by centrifugation and immediately frozen on dry ice. Samples were stored at approximately −80°C until analysis.
Analytical methodology for SB 207499.
Plasma concentrations of SB 207499 were determined after a liquid-liquid extraction procedure, using a validated GC method. To 100 μl of plasma were added 25 μl of internal standard and 1 ml of acetic acid (0.1 N). Five ml of hexane:methyl-t-butyl ether (1:1) was added and the mixture was shaken for 30 min, then centrifuged for 10 min to separate the layers. The organic layer was removed and evaporated to dryness under a stream of nitrogen at −55°C. Derivatization was accomplished by adding methanolic HCl (200 μl) to the residue and heating at −95°C for 15 min. The solution was then evaporated to dryness under a stream of nitrogen at −55°C, and the residue was reconstituted in 50 μl of toluene prior to GC analysis. Five microliters of the reconstituted solution was injected onto the gas chromatograph (HP 5880A) in the split injection mode (split ratio 20:1) using a thermionic detector. Chromatographic analysis was accomplished using a 30 m DB-1 wide bore column (0.25 U, 0.32 mm id) (J&W) under the following conditions: column temp 250°C (isothermal); injector temperature 250°C and detector temp 300°C. The assay was linear over the concentration range 10 to 10,000 ng/ml, using a 100-μl plasma sample.
Tolerance induction study.
Balb/c male mice in groups of three to eight were treated with test compound for 4 days (8 mg/kg/day, p.o. −1.5 to 2 × ED50) or once 30 min before the injection of 2 to 10 × 106 human monocytes per ml into the peritoneal cavity. The animals were then treated as described in the methods section entitled, “In vivo administration of human monocytes for the production of TNFα.”
Mouse endotoxin-induced shock.
Mice (three to five group) received an oral dose of SB 207499 30 min before an intravenous challenge with LPS (0.1 μg) and d-galactosamine (500 mg/kg). Serum was harvested 1 hr after challenge and mouse TNFα levels were measured by ELISA as described above.
Oxazolone-induced contact sensitivity.
Mice were sensitized by a single application of 10 μl of a 1.6% solution of oxazolone (Sigma) in ethanol on the left ear. To induce a chronic lesion, the mice were challenged with 10 μl of a 0.8% solution of oxazolone three times per week for 4 wk postsensitization (mice challenged days 7, 9, 11, 14, 16, 18, 21, 23, 25, 28, 30 and 32 after sensitization). Ear thickness was measured with a dial micrometer (Mitutoyo, Japan) and tissue samples collected 24 hr after the last challenge over a 24-hr period.
Measurement of IL-4 by specific ELISA.
Individual mouse ears from the experiments were homogenized in 1 ml of phosphate-buffered saline (pH 7.4) and assayed for IL-4 and by specific ELISA (INTERTEST-4X ELISA kits Genzyme Diagnostics, Cambridge, MA). The assays were performed as recommended by the manufacturer.
Compounds.
SB 207499 [c-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)-r-l-cylclohexanecarboxylic acid], (R,S and R/S)rolipram [4-(3-cyclopentyloxy-4-methoxyphenyl)-2-(1H)-pyrrolidone], denbufylline [1H-purine-2,6-dione,1,3-dibutyl-3,7-dihydro-7-(2-oxopropyl)] and tibenelast (LY-186,655, benzo-[b] thiophene-2-carboxylic acid, 5,6-diethoxy), CP 80633 [5-(3-exo-bicyclo[2.2.1]-hept-2-yloxy-4-methoxyphenyl)-3,4,5,6-tetrahydro-pyrimidin-2-(1H)-one] were synthesized or obtained by Siegfried Christensen and colleagues, Department of Medicinal Chemistry, SmithKline Beecham Pharmaceuticals (King of Prussia, PA). Polyethyleneglycol 200 (PEG200) purchased from Sigma or olive oil were used as vehicles. Reserpine was obtained from Novartis Pharmaceuticals, Summit, NJ.
Data analysis.
All data are presented as mean ± S.E. Statistical analysis was performed using Student’s t test and P <.05 was considered statistically significant relative to vehicle control. ED50s were calculated using regression or probit analysis.
Results
To establish further the role of PDE4 in the regulation of TNFα production in vivo, the activities of the first generation PDE4 inhibitors rolipram, denbufylline and tibenelast (LY-186,655) were examined in the adoptive transfer model and compared to SB 207499. As seen in table 1, all compounds, given at a dose of 50 mg/kg, p.o., significantly inhibited the production of both human and mouse TNFα (70–81% inhibition), although tibenelast produced more modest inhibition, particularly of mouse TNFα production (38% inhibition). In addition (data not shown), S-rolipram, the less active enantiomer with respect to PDE4 inhibition, did not inhibit human TNFα production and had only limited action on mouse TNFα production (42%; P <.01) at a dose of 50 mg/kg, p.o. Taken together, these results further underscore the conclusion that PDE4 inhibitors have the capacity to inhibit TNFα production in thisin vivo model.
Effect of selected PDE4 inhibitors on human and mouse TNFα production in vivo
It was of interest to examine in more detail the potency of SB 207499 in comparison to R-rolipram. Dose-related inhibition of human TNFα production by SB 207499 and R-rolipram was clearly demonstrable in this model. As seen in figure 1, the administration of SB 207499 or R-rolipram to Balb/c mice just after the injection of human monocytes and before LPS challenge strongly inhibited the production of human TNFα in a dose-dependent fashion, with ED50s of 4.9 and 5.1 mg/kg, p.o., respectively.

Inhibition of human TNFα by orally administered SB 207499 (A) or R-rolipram (B). Balb/c mice (n = 4–7/group) were injected with human monocytes as described, drug or vehicle was orally administered at the doses indicated and LPS was injected i.p. Two hr later, the animals were euthanized and a peritoneal washout obtained for subsequent ELISA analysis. The dose-response shown is mean ± S.E. percent inhibition and the ED50 calculated by regression analysis.
To compare the CNS activity of R-rolipram and SB 207499, the ability of these compounds to reverse reserpine-induced hypothermia was evaluated in Balb/c mice. Whereas, SB 207499 and R-rolipram were equipotent as inhibitors of LPS-induced TNFα production, SB 207499 was 10-fold less potent than R-rolipram in this standard antidepressant model (fig.2).

Reversal of reserpine-induced hypothermia by R-rolipram (○) or SB 207499 (•). Balb/c mice (n = 8–9/group) were individually housed and baseline rectal temperatures obtained. The mice were then given reserpine (10 mg/kg, i.p.). Four hours later, the rectal temperatures were again recorded and test compound or vehicle was administered by gavage. Ninety minutes later, the rectal temperature was measured and dose-response curves were constructed. ED50s were determined by probit analysis.
The duration of action of SB 207499 was also assessed. As seen in figure 3, the duration of action of orally administered SB 207499 (30 mg/kg) against mouse TNFα production was at least 10 hr, indicating a long duration of action in the mouse. Plasma samples from these animals were also taken to determine whether the prolonged duration of action of SB 207499 coincided with a prolonged elevation of drug plasma concentrations. Indeed, the plasma concentrations of SB 207499 (30 mg/kg, p.o.) were as great as 6491 ± 371 (15 min) and ranged from 165 to 500 ng/ml (ca. 0.5–1.5 μM) over a period of 4 to 10 hr (fig. 3).
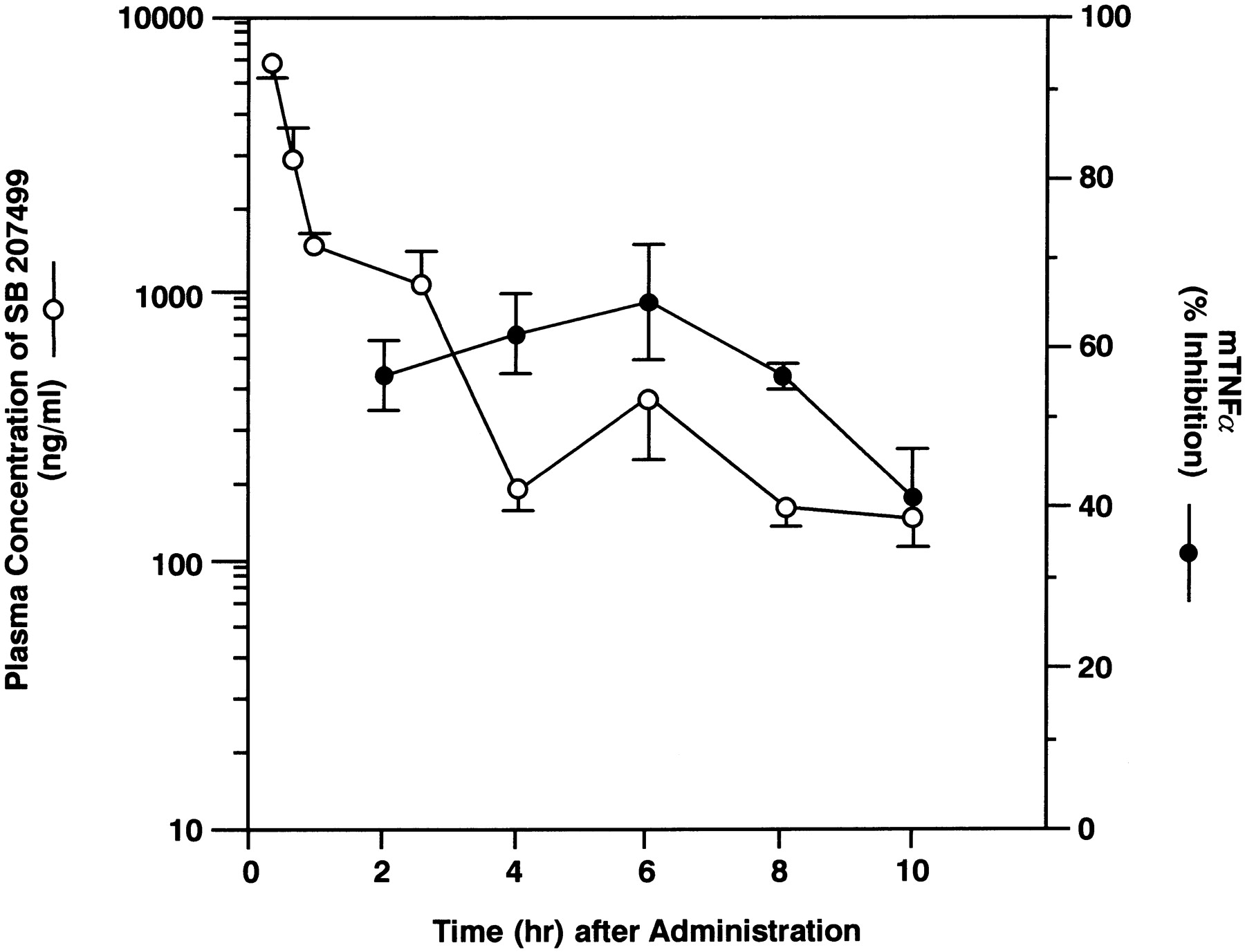
Pharmacokinetic/pharmacodynamic relationship for SB 207499. Balb/c mice (n = 3–5/group) were administered SB 207499 (30 mg/kg, p.o.). Animals were euthanized and blood samples taken at the intervals shown. Plasma concentrations of drug (○) were determined as described (shown as mean ± S.E.). Samples for measurement of TNFα production (•) were taken beginning 2 hr postdrug administration. LPS challenge was administered 15 min after drug administration. Data shown are mean ± S.E. Baseline value for TNFα in vehicle-treated and LPS challenged mice was 635.1 ± 60.3 pg/ml.
Tolerance to SB 207499 was not observed after repeated administration. As seen in figure 4, activity after the administration of four daily doses (1.5–2 × ED50) of either vehicle, R-rolipram or SB 207499, was compared to the activity of these treatments given once. In the case of SB 207499, inhibition of human TNFα production improved (44 vs. 75% inhibition) after multiple treatment. In contrast, R-rolipram appeared to be less active after 4 days of administration (44 vs. 24% inhibition). Also seen in figure 4 is a similar pattern for the inhibition of mouse TNFα.

Evaluation of tolerance upon multiple dosing of SB 207499 or R-rolipram. Balb/c mice (n = 5–8/group) were given either a single dose of R-rolipram (10 mg/kg, p.o.) or SB 207499 (8 mg/kg, p.o.), or four daily doses of each compound. In all cases, the last doses where administered 30 min before injection of human monocytes and LPS challenge. Two hours postchallenge, animals were euthanized and peritoneal washouts obtained for TNFα analysis as described. Data shown are the mean ± S.E. of the TNFα concentrations (pg/ml). * Statistically different from the vehicle control, P <.05.
In addition to demonstrating inhibition of TNFα production in peritoneal washouts, the activity of SB 207499 against the generation of TNFα in blood was demonstrated in a model of endotoxin-induced shock. In this model, C57B1/6 mice were administeredd-galactosamine along with the i.v. challenge with LPS. Oral administration of SB 207499 at doses of markedly reduced the serum TNFα levels in a dose-related fashion (fig.5). The ED50 was calculated to be 7.7 mg/kg, p.o., virtually identical to the value obtained using peritoneal TNFα as a marker for activity.

Effect of SB 207499 on TNFα levels ind-galactosamine, LPS-induced endotoxic shock. Balb/c mice (n = 3–5/group) were given test compound or vehicle 30 min prior to i.v. challenge with LPS (0.1 μg) andd-galactosamine (500 mg/kg). Serum was harvested 1 hr after challenge and TNFα levels were measured as described. Data shown are mean ± S.E. * Statistically different from the vehicle control, P < .05.
IL-4 plays an important role particularly in allergic disease (Crockeret al., 1996). It was of interest, therefore, to examine the ability of SB 207499 to inhibit IL-4 production in vivo. To achieve this, chronic oxazolone-induced contact sensitivity in mouse skin was used (Webb EF, et al., 1998). As seen in table 2, SB 207499, rolipram and CP 80633 were active in this model. The inflammatory response to oxazolone and intralesional IL-4 concentrations were significantly inhibited by topical SB 207499, rolipram and CP 80633. That the activity was limited to PDE4 inhibitors was suggested further by the results of evaluation of inhibitors of other PDE isozymes including the PDE1 inhibitor, vinpocetine, the PDE3 inhibitor, siguazodan and the PDE5 inhibitor, zaprinast. None of these compounds demonstrated inhibitory activity in this model system (data not shown).
The topical effects of PDE4 inhibitors on chronic oxazolone-induced contact sensitivity
Discussion
These results demonstrate the ability of SB 207499 to reduce inflammatory cytokine production in vivo. This cytokine modulatory activity is shared by other PDE4 inhibitors including, rolipram, denbufylline and tibenelast. It is of interest to note that with the exception of tibenelast, these compounds inhibited both human and mouse TNFα production with essentially equal potency. Although the caveat of different cell sources for TNFα (monocyte for human and liver for mouse; Luster et al., 1994) applies, these results suggest that the activity of PDE4 inhibitors in the mouse LPS-induced TNFα model might predict similar activity in humans. Further support comes from the observation that even the relatively nonspecific but clinically used agent, theophylline can be shown to inhibit TNFα production in the rat, although SB 207499 was found to be ∼25 times more potent (Griswold DE, unpublished results). That the effects observed are attributable to SB 207499 itself is suggested by thein vitro studies showing inhibition of TNFα production from human monocytes (Barnette et al., 1998) and by the lack of major circulating metabolites in the rat or the monkey (data not shown).
To evaluate the effect of SB 207499 on the systemic production of TNFα, the i.v. challenge with LPS in mice treated withd-galactosamine where extremely high, sometimes lethal levels of TNFα are seen, was used. The administration of SB 207499 at doses from 3 to 50 mg/kg, p.o. dramatically reduced the systemic concentration of TNFα with an ED50 of 7.7 mg/kg, p.o. These data provide yet another perhaps more stringent setting where the inhibition of inflammatory cytokines by PDE4 inhibitors is apparent.
An additional model that was used to evaluate the effect of SB 207499 on IL-4 production was oxazolone-induced contact sensitivity. Chronic T cell activation in this model leads to a switching of Th1- to Th2-mediated events and results in a pattern of cytokine generation consistent with that observed in allergic disorders such as asthma and atopic dermatitis (Webb et al., 1998). This includes a dramatic upswing in IL-4 production in the lesional skin. Topical administration of SB 207499, but not inhibitors of PDE1, PDE3 or PDE5, inhibited IL-4 production in this lesion. It thus shares this activity with CP 80633 which has been shown to be clinically effective as a topical treatment for atopic dermatitis, a skin disease strongly associated with IL-4 production (Hanifin et al., 1996). It is intriguing that the acute oxazolone response, typified by interferon-γ production was relatively insensitive to SB 207499 (data not shown). Taken together, these results suggest that the Th-2-associated responses may be most appropriate for SB 207499 and PDE4 therapy. It is unknown at present whether or not these effects extend to inhibition of CD23 expression or IgE synthesis. None of the PDE4 inhibitors were active given orally. This probably reflects the typically poor penetration of compounds into the cutaneous compartment from the vasculature.
In addition, SB 207499 has other favorable characteristics. In particular, SB 207499 has a long duration of action with significant inhibition of LPS-induced TNFα production demonstrable for at least 10 hr after drug administration. Also, unlike rolipram, the multiple dose studies revealed no diminution of activity after dosing for 4 days. Thus no evidence of either pharmacological tolerance or induction of metabolism was demonstrable.
Enthusiasm over the potential use of PDE4 inhibitors as antiinflammatory agents is tempered by concern over class-associated side effects. These side effects include increased gastric acid secretion (Barnette et al., 1995), nausea and vomiting (Horowski and Sastre-Y-Hernandez, 1985). Furthermore, the first generation PDE4 inhibitor, rolipram, was originally developed as an antidepressant agent (Horowski and Sastre-Y-Hernandez, 1985), indicating that it possesses psychotropic activity. Thus the challenge to drug discovery has been to design novel, second generation PDE4 inhibitors that maintain the therapeutic activity of older compounds, but have a reduced potential to elicit side effects. One approach toward the rational design of such compounds hinges on the existence of two distinct conformers of PDE4 (Torphy et al., 1992;Jacobitz et al., 1996). One of these conformers, termed “high affinity rolipram-binding PDE4” or HPDE4, is inhibited by nanomolar concentrations of rolipram. The second conformer, termed “low affinity rolipram-binding PDE4,” is 10- to 100-fold less sensitive to rolipram. Other first generation PDE4 inhibitors display a similar preference for HPDE4 (Barnette et al., 1997; Souness and Rao, 1997). Importantly, HPDE4 appears to predominate in the CNS (Saccomano et al., 1991) and in parietal glands (Barnetteet al., 1995), whereas LPDE4 predominates in many inflammatory cells (Barnette et al., 1997; Souness et al., 1996), including human monocytes (Barnette et al., 1996; Souness et al., 1996). Collectively, this information suggests that an improved therapeutic ratio is attainable by synthesizing compounds with a decreased relative activity against HPDE4. Indeed, SB 207499 is a second generation PDE4 inhibitor specifically designed on the basis of these principles. SB 207499 is equipotent to R-rolipram against LPDE4 (IC50 ≅ 100 nM), whereas its potency against HPDE4 (IC50 = 120 nM) is 60-fold less than that of R-rolipram (IC50 = 2 nM) (Torphyet al., 1997; Christensen et al., 1998).
To provide support for the concept that decreasing the relative activity of a compound against HPDE4 results in an improved therapeutic ratio, we determined the relative potencies of SB 207499 and R-rolipram for inhibition of TNFα production vs. reversal of reserpine-induced hypothermia in Balb/c mice. The latter model provides a general index of CNS effects and is predictive of antidepressant activity (Sanghvi and Gershon, 1977). In our study, R-rolipram and SB 207499 were equipotent in suppressing TNFα production, but SB 207499 was 10-fold less potent as a CNS-active agent. This outcome is reminiscent of a previous study in which SB 207499 was equipotent to R-rolipram as an inhibitor of TNFα production from isolated monocytes, but 150-fold less potent as an acid secretagogue in vitro. Taken together, this information supports the concept that the side effect profile of SB 207499 will be improved compared with first generation PDE4 inhibitors.
In summary, these results indicate that SB 207499 is a potent, orally active inhibitor of cytokine production. Moreover, SB 207499 has a long duration of action and possesses an improved therapeutic ratio compared with R-rolipram. Thus, SB 207499 is an excellent drug candidate to test the therapeutic value of PDE4 inhibition in inflammatory disease. Indeed, SB 207499 is currently in phase II clinical trials for the treatment of asthma.
Footnotes
-
Send reprint requests to: Dr. Don E. Griswold, Associate Director, Department of Pulmonary Pharmacology, SmithKline Beecham Pharmaceuticals, 709 Swedeland Road, P.O. Box 1539, King of Prussia, PA 19406-0939.
- Abbreviations:
- Cyclic AMP
- 3′-5′cyclic adenosine monophosphate
- CNS
- central nervous system
- DPBS
- Dulbecco’s phosphate-bufferred saline without calcium and magnesium
- ELISA
- enzyme-linked immunosorbant assay
- HPDE4
- PDE4 conformer that binds rolipram with high affinity (previously termed “high affinity rolipram binding site”)
- LPS
- lipopolysaccharide
- LPDE4
- PDE4 conformer that binds rolipram with low affinity
- PDE4
- phosphodiesterase type 4
- TNFα
- tumor necrosis factor α
- IL-4
- interleukin-4
- Received February 18, 1998.
- Accepted June 26, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}