Article Text

Abstract
A patient is described with a novel syndrome characterised by progressive muscular weakness, contractures, pupillary muscle dysfunction, and skeletal deformity. The main myopathological feature was an abundance of tubular aggregates in both type I and type II muscle fibres. Myopathies in which tubular aggregates are the defining feature are rare and either present with progressive muscle weakness or exercise induced myalgia. Tubular aggregate myopathy with symptomatic smooth muscle dysfunction and skeletal deformities has not been described before.
- tubular aggregates
- pupil
- myopathy
Statistics from Altmetric.com
Tubular aggregates are inclusions within muscle fibres formed from the terminal cisterns of the sarcoplasmic reticulum.1 They are observed in a range of specific neuromuscular conditions in which they are the main myopathic feature, including familial limb girdle myasthenia,1–4 exercise induced myalgia,1,5–10 and gyrate atrophy of the choroid and retina.11 They are also found in various other disorders in which they are inconsistently present and are often a minor part of the overall myopathology. These include periodic paralysis,12–14 paramyotonia,15 and metabolic myopathies (notably ethanol induced myopathy16–18). In addition, they may be features in patients with idiopathic and congenital myopathies.19 The pathological significance of tubular aggregates is unknown; however, an attractive hypothesis is that they are a response of the sarcoplasmic reticulum to injuries affecting excitation–contraction coupling or calcium flux.1
We describe a patient with a novel tubular aggregate myopathy characterised by progressive weakness with prominent contractures, pupillary muscle involvement, and skeletal abnormalities.
CASE REPORT
A 51 year old man presented with weakness in his arms and legs developing over four years. He was born a normal full term delivery and had normal early motor milestones. He reported that he had always tired of physical work more quickly than his peers and that he had avoided sport from childhood, as he had always become rapidly fatigued. Over the last four years he had noticed increasing difficulty in walking, lifting heavy objects, and manipulating objects in his hands. The weakness was associated with aching in his muscles. He had dysphagia for solids and there had been a change in the quality of his voice. In addition, he reported difficulties with night vision.
Of his four siblings, two had developed arthritis in their third decade, but there was no family history of neuromuscular disease. At presentation, he consumed less than six units of alcohol a week. There was no history of exposure to myotoxic drugs.
Examination
General physical examination revealed a tall man (1.96 m) with a mild saddle nose deformity and pectus excavatum (fig 1). There were no clinical features of a connective tissue disease. He could tandem walk with difficulty but was unable to walk on tiptoes or on his heels. He was unable to stand from sitting without using his arms.
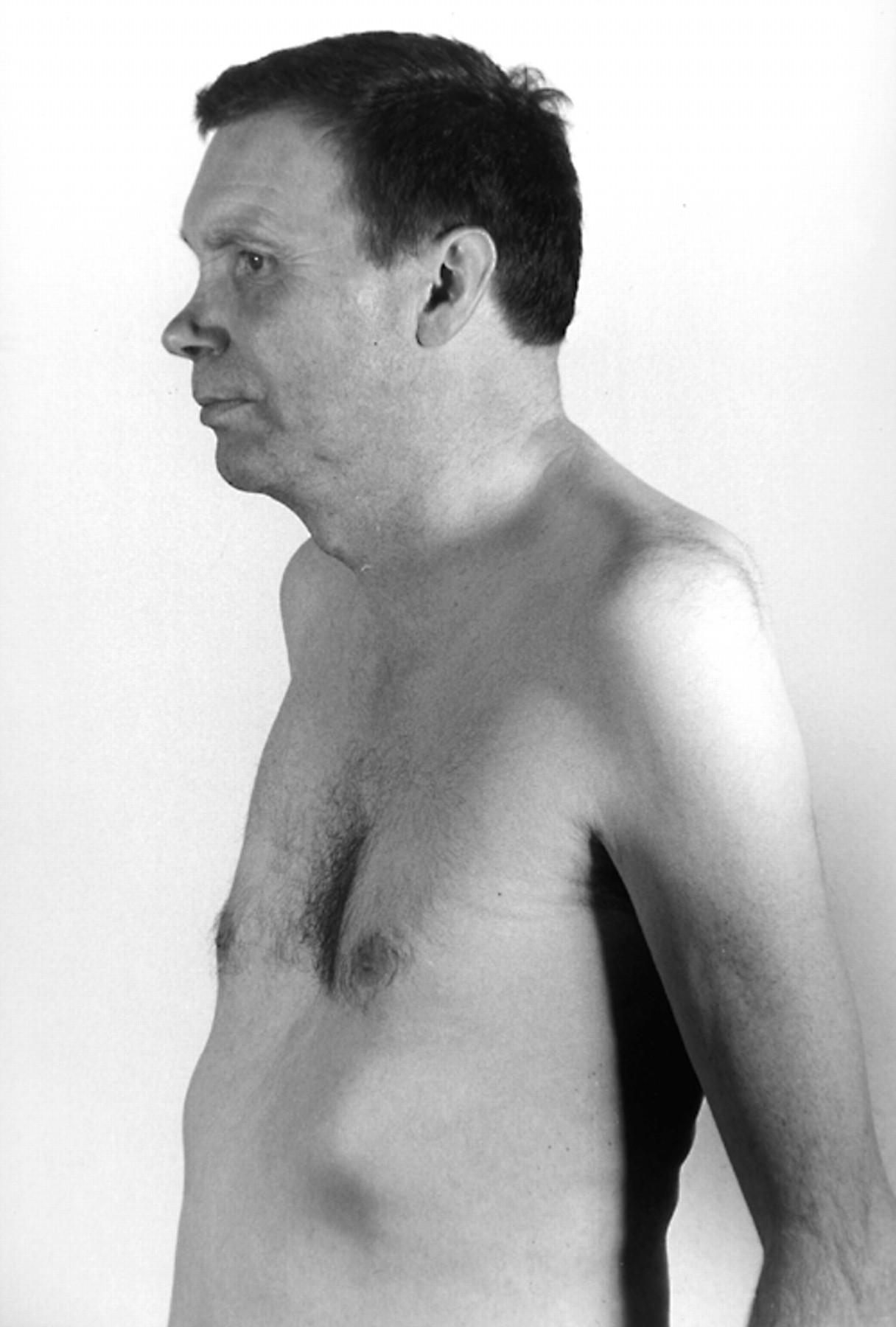
The patient had various skeletal abnormalities including pectus excavatum and a mild saddle nose. Proximal muscle wasting is also visible.
His uncorrected visual acuity was 6/6 in both eyes. Both pupils were circular, regular in outline, equal in size, but markedly constricted (1.5 mm) in the dark. On slit lamp examination, there were small but brisk responses to light and near. There was no pupillotonia, no segmental palsy, and no light-near dissociation. Instillation of tropicamide 1% plus phenylephrine 10% dilated the pupils to only 2.5 mm. The fundi and external ocular movements were normal.
There was a minor degree of wasting of the temporalis and masseter muscles on both sides. There was symmetrical wasting and weakness of the sternomastoid and trapezeus muscles, with grade 4/5 weakness on the MRC scale. Examination of the cranial nerves, including the muscles of facial expression and the tongue, was otherwise unremarkable. Contractures affecting the neck extensors limited neck flexion.
There was symmetrical wasting of both proximal and distal muscles in the upper and lower limbs, particularly involving the periscapular muscles, the flexor compartment of the forearms, and the quadriceps femori. There were contractures that limited finger and wrist extension. There was symmetrical proximal and distal weakness throughout the upper and lower limbs of grade 4/5 and 4+/5 on the MRC scale. The biceps, triceps, supinator, and ankle reflexes were absent on both sides. The knee jerks were absent on the right and present with reinforcement on the left. Plantar reflexes were flexor. Sensory examination was normal.
Investigations
Routine biochemistry was normal, including tests of renal, liver, and thyroid function, and plasma calcium and phosphate. The erythrocyte sedimentation rate was 7 mm/hour (normal range 1–20) and C reactive protein was 15.3 mg/l (normal range < 10). Serum creatine kinase ranged between 376 and 3854 IU/l (normal range < 150). A random venous lactate concentration was normal. Screening for autoantibodies including antinuclear antibody, rheumatoid factor, and Jo-1 was negative.
Electromyography revealed a patchy chronic myopathic picture affecting particularly the wrist flexors and characterised by the presence of excess polyphasic units. Repetitive stimulation excluded a disorder of the neuromuscular junction, and there was no clear evidence of a neuropathy, neither was there evidence of cardiomyopathy on electrocardiography or echocardiography.
A muscle biopsy was taken from the vastus lateralis. Frozen sections were cut and stained using tinctorial and enzyme histochemical techniques according to established protocols, and examined by light microscopy. Glutaraldehyde fixed tissue was processed and stained for ultrastructural examination. Light microscopy showed increased variation in fibre size with fibre diameters predominantly in the range 90–200 μm and with interspersed atrophic fibres. The most conspicuous feature was the presence of peripheral and central inclusions of granular material in many type I and type II fibres; this material was basophilic on haematoxylin and eosin staining and red on staining with the Gomori trichrome method. The inclusions had the characteristics of tubular aggregates, showing strong activity on nicotinamide adenine dinucleotide dehydrogenase tetrazolium reductase (NADH-TR) histochemistry and marked myoadenylate deaminase activity, but neither succinic dehydrogenase (SDH) nor cytochrome oxidase (COX) activity (fig 2). There was a considerable increase in interstitial connective tissue. Split fibres were present, internal nuclei were frequent, and there were small numbers of actively regenerating fibres. No inflammation or fibre necrosis was seen. Electron microscopy confirmed the presence of tubular aggregates (fig 2).

{kind=link}
{kind=link}
Frozen sections from vastus lateralis examined by light microscopy showed granular inclusions located both centrally and beneath the sarcolemma of myofibres. (A) Haematoxylin and eosin (bar = 80 μm). (B) Myoadenylate deaminase (bar = 50 μm). (C) NADH-TR (bar = 80 μm). (D) Electron microscopy confirms the presence of tubular aggregates within myofibres (bar = 300 nm).
DISCUSSION
We describe a patient who had a novel myopathic syndrome with tubular aggregates. The distinctive features in our case are prominent contractures, skeletal deformity, and abnormal pupils.
While there are many causes of tubular aggregates (for example, periodic paralysis, paramyotonia congenita, familial myasthenia, gyrate atrophy of the retina and choroid, and metabolic insults1–4,12–18), we can exclude all these in our patient on the basis of the clinical history and routine investigations. In addition, there is a group of patients with prominent symptoms of exercise induced myalgia and fatigue in whom the principal pathological finding is numerous intramuscular tubular aggregates.5–11 Our patient gave a history of reduced exercise tolerance from childhood, but the prominent and progressive weakness, contractures, ocular involvement, and skeletal abnormalities distinguished him from that group of patients.
Tubular aggregates have also been described in mouse models of autoimmune connective tissue diseases20 and have been described rarely in polymyositis in humans.21 Although our patient had a family history of arthritis, he had no clinical, biochemical, or serological evidence of a connective tissue disorder. Furthermore, there was no histological evidence of inflammatory muscle disease.
There have been reports of several patients with idiopathic tubular aggregate myopathies, without the other features of the syndromes mentioned above. These can occur as sporadic cases22 or as part of familial syndromes.19 In some of the familial cases, tubular aggregates occur in both type I and type II muscle fibres. In contrast, most other patients with tubular aggregate myopathies—including those with prominent exercise induced myalgia—have tubular aggregates restricted to type II fibres only.23 In this respect, our patient resembles the rarer familial cases. While these cases have a variable range of clinical features we are unaware of a description of a case with the same combination of musculoskeletal and pupillary features found in ours.
An interesting feature of our patient was the marked bilateral pupil abnormality. The extreme degree of meiosis suggested that these pupils may be either Argyll-Robertson or old Holmes-Adie pupils, but such descriptions are inappropriate because of the regularity of the pupillary outline, the presence of brisk, though inevitably small, light reflexes, and the lack of light-near dissociation. Could the meiosis be the result of a sympathetic deficit such as occurs in Horner’s syndrome? Two observations make this unlikely: the pupils were too small for this condition, and they did not dilate with phenylephrine. Taken together, the presence of reflex activity and the failure of phenylephrine mydriasis indicate that the abnormality is unlikely to be neuropathic in origin. In the circumstances, we are inclined to believe that it arises from a myopathy of the iris musculature, particularly the pupil dilator.
Abnormalities of pupillary smooth muscle have been described in association with a tubular aggregate myopathy in a small number of patients with gyrate atrophy of the choroid and retina. These patients have a deficiency of ornithine transaminase and present with progressive blindness caused by photoreceptor and choroid degeneration.11 Symptoms of skeletal muscle disease are uncommon in such patients, but many have tubular aggregates in their skeletal muscle. Although these patients have normal pupillary function, samples of the pupil dilator muscle contain structures resembling tubular aggregates.24 This is surprising as tubular aggregates are thought to arise from the sarcoplasmic reticulum, a structure which is not present in smooth muscle. Either the tubular aggregate-like structures seen in the iridectomy samples are completely different entities from those seen in skeletal muscle, or tubular aggregates can represent a more generalised pathological feature of intracellular calcium stores.
In contrast, our patient presented with widespread skeletal muscle disease and symptoms and signs suggestive of pupillary muscle dysfunction but with normal visual acuity and fundi. We hypothesise that he has a tubular aggregate myopathy affecting his pupillary smooth muscle in addition to his skeletal muscle.