





Abstract
Airway inflammation is now recognized as a major factor in the pathogenesis of lung disease in cystic fibrosis (CF). Its most characteristic feature is a marked and persistent influx into the airways of neutrophils, which damage the lung by releasing noxious mediators, such as reactive oxygen species and proteolytic enzymes.
Recent studies suggest that inflammation occurs very early and may even happen in the absence of infection. Furthermore, links between CF transmembrane conductance regulator dysfunction and both infection and inflammation are postulated; dysregulation of cytokine production and abnormal epithelial host defences have been regarded as causes of sustained inflammation.
Bronchoalveolar lavage and the evaluation of neutrophils and inflammatory mediators provide the most accurate picture of airway inflammation. Routine bronchoscopy with bronchoalveolar lavage, however, is unpleasant for the patient and usually is of no immediate benefit to the management of individual cases.
Therefore, surrogate markers collected by noninvasive procedures would be of great assistance in the follow-up of cystic fibrosis patients. Several markers have been evaluated in the sputum, serum and urine of cystic fibrosis patients and related to the degree of airway inflammation. Long-term studies are needed to confirm their potential clinical utility and specificity, and to determine which can be used clinically to monitor disease outcome and efficacy of treatment.
Cystic fibrosis (CF) is the most common life-shortening, autosomal, recessive disorder in Caucasian populations. It is caused by mutations in a single gene, located on the long arm of chromosome 7, which encodes a 1,480 residue transmembrane glycoprotein. This is the CF transmembrane conductance regulator (CFTR) 1, which acts as a cyclic adenosine monophosphate-regulated chloride channel at the apical membrane of epithelial cells. Recent data indicate that CFTR is also involved in the regulation of other ion channels (either stimulating other chloride channels or inhibiting the epithelial sodium channel), endosome fusion and intravesicular trafficking 2. CFTR is defective, mislocalized or absent in CF 1, leading to perturbed transport of salt and water across epithelial barriers. CF is characterized by a wide variability of clinical expression; dehydration and plugging of mucous secretions in the ducts of exocrine glands predispose to multi-organ clinical manifestations, particularly within the gastrointestinal, hepatobiliary, reproductive and respiratory tracts. The main clinical features may include chronic obstructive pulmonary disease, exocrine pancreatic insufficiency, high content of Na+ and Cl− in sweat, and male infertility.
Progressive lung disease characterized by chronic airway infection with specific flora is the prime cause of morbidity and mortality in CF patients 1. Despite recent advances in understanding the cellular and molecular basis of CF, the sequence of events leading to irreversible lung damage is still poorly defined. Airway inflammation, however, is now recognized as a major factor 3, 4. The traditional concept is that a primary genetic defect causes defective chloride secretion and increased sodium re-absorption, leading to hyperviscous secretions within the airways, thereby impairing mucociliary clearance. Chronic bacterial infection is favoured and induces abnormal and persistent inflammation. However, recent studies have suggested that inflammation is a very early event and may even occur in the absence of infection. Indeed, a direct link between defective CFTR and both infection and inflammation may exist 3. Whatever their temporal relationship, infection and inflammation become intimately linked early in the course of the disease, with each exacerbating the other. Thus, a vicious circle of infection and inflammation is established that is mainly responsible for progressive and irreversible lung damage. This review will focus on the most recent advances in knowledge about the onset and aetiology of inflammation in CF lung disease, its role in lung damage and its relationship with airway infection. Inflammatory response markers in CF will also be described.
Cystic fibrosis lung disease
The main clinical features of CF lung disease are chronic airway infection and repeated acute exacerbations, with distinctive bacterial flora precipitating irreversible lung damage. The CF lung is normal in utero and during the first few months of life, except for mild dilation of tracheal submucosal glands 5, 6. Shortly after birth, however, many patients become infected with bacteria and develop airway inflammation. Plugging of the small airways by abnormal mucus is the most prominent early manifestation, accompanied by inflammatory infiltrates in the mucosa and submucosa. With time, excessive mucus production, airway obstruction, chronic airway infection and chronic inflammation in the airways lead to severe bronchiectasis and respiratory failure.
Micro-organisms responsible for airway infection in cystic fibrosis
Although many micro-organisms are capable of infecting the CF lung, the major pathogens are the bacteria that chronically infect the airways: Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa. S. aureus and H. influenzae inhabit the CF airway early, often before the onset of clinical symptoms, whereas infection with P. aeruginosa almost universally follows the other pathogens 7, 8. There is a special association between CF and P. aeruginosa; chronic infection of the airways with this micro-organism, particularly mucoid strains, is closely associated with progressive pulmonary deterioration, although the rate of progression is highly variable. Most patients become chronically infected during school age or early adolescence. Early colonization with nonmucoid strains can sometimes be eradicated, but strains tend to persist and alter their phenotype. The emergence of the alginate-producing mucoid variants, highly characteristic of CF, is associated with a more rapid decline in lung function. Once established within the CF airways, P. aeruginosa is notoriously resistant to eradication by chemotherapy 1, 8–10. It chronically colonizes the bronchial lumen and virtually never breaches the epithelial barrier.
In the past decade, infection with Burkholderia cepacia has become an increasing concern. Strains of this multiresistant organism can also infect the CF airways and some demonstrate direct cross-infection between patients. Chronic B. cepacia infection is often accompanied by an adverse clinical course (rapid deterioration with bacteriaemia and death is seen in 20–30% of infected patients) 8, 10.
Susceptibility to airway infection in cystic fibrosis
The causal association between the basic defect and susceptibility to airway infection in CF is a timely but highly controversial area of research. No systemic immune defect that would account for the peculiar susceptibility of CF patients to infection with specific flora has been documented. Increased infection outside the respiratory tract has not been observed. Bacteria remain primarily endobronchial and do not cause the invasive or disseminated infections noted in immunocompromised hosts 1, 4, 10. These clinical data suggest that CF lung disease is initiated by a breach of the host defence system of the airways and propagated by an inability to effectively clear the infection. A number of hypotheses have been proposed to explain how CFTR dysfunction causes CF patients' susceptibility to airway infection. Using cultured epithelial cells from freshly excised bronchial specimens, Matsui et al. 11 found that neither Na+ and Cl− concentrations nor the osmolarity of surface fluid differ in normal and CF cultures. However, CF epithelia display accelerated isotonic fluid absorption that depletes airway surface fluid volume, leading to impaired mucociliary clearance and thickened mucus. These findings support the traditional view that a major defect in CF is mucus dehydration, impairing mucociliary clearance and favouring airway infection by CF-associated pathogens. Studies of mucus from CF patients suggest a spectrum of rheological abnormalities 12, whereas in vivo mucociliary clearance studies are inconclusive 13, 14. Furthermore, comparison of the clinical course and impairment of mucociliary clearance between CF and the primary ciliary dyskinesia syndrome strongly suggests that a clearance defect is not enough to account for the increased susceptibility to infection and the progressive obstructive lung disease observed in CF. In the primary ciliary dyskinesia syndrome, mucociliary clearance is more severely impaired than in CF, whereas lung disease is much less severe and chronic airway infection with P. aeruginosa is less frequent and occurs much later in the course of the disease 15.
In contrast, alternative hypotheses suggest that defects in CFTR result in elevated salt concentrations that compromise innate defence mechanisms within the CF airway surface fluid (ASF). ASF contains a variety of interacting antimicrobial factors including antibacterial peptides and β-defensins 1 and 2, which kill P. aeruginosa and other species 16–18. The activity of these peptides is salt-sensitive and markedly reduced in the ASF of cultures of airway epithelial cells from CF patients. The antibacterial activity of CF ASF can be restored by lowering its salt concentration in vitro. This suggests that the components of the defence system are intact but unable to function correctly in the abnormally high NaCl concentrations that may be found in the airway lumen of CF patients 16. At the core of this theory is the elevated NaCl content in CF ASF, an observation that is controversial 19. Additional studies and the development of techniques to provide accurate measurements of the electrolyte composition of CF ASF in vitro and in vivo may confirm this hypothesis.
Recent evidence suggests that the interaction between airway epithelial cells and bacteria is impaired in CF as a consequence of CFTR dysfunction. These abnormalities may therefore directly interfere with host defence.
In vitro studies show that P. aeruginosa adheres more to respiratory epithelial cells from CF patients and particularly to damaged and regenerating cells than to control cells 20, 21. This is thought to be due to an alteration in the number of receptors for P. aeruginosa adhesins on the cell surface, which in turn is influenced by CFTR. Indeed, P. aeruginosa pilin, the receptor responsible for P. aeruginosa binding, recognizes and binds to the cell surface glycolipid asialo ganglioside M1 (aGM1), but not to the sialylated homologues. aGM1 is present in higher levels in CF airway epithelial cells as a consequence of CFTR dysfunction 20.
CFTR has also been implicated in the uptake of P. aeruginosa by cultured human airway epithelial cells. It has been shown that cells expressing the ΔF508 allele of CFTR are defective in the uptake of bacteria 22. Although the clinical relevance of this in vitro observation is still unclear, this mechanism might further contribute to the impaired clearance of micro-organisms in CF. Taken together, these observations suggest a direct link between the primary defect of CFTR and the impairment of local host defences in CF.
Whatever the mechanisms of initiation of infection, once established, chronic P. aeruginosa infection is critical in accelerating the progression of lung disease in most CF patients. Bacteria persist in the airways despite a strong host response, reflected by the production of antibodies to many P. aeruginosa antigens and the formation of immune complexes 4, 23. A phenotype switch by P. aeruginosa to mucoid growth in macrocolonies that inhibit phagocytosis, is thought to be the main persistence mechanism. The highly antigenic mucoid exopolysaccharide produced by P. aeruginosa leads to the formation of immune complexes in situ, which further stimulate the inflammatory response. This inflammatory response prevents the systemic spread of infection, but fails to eradicate it from the airways, ultimately causing irreversible damage to the airways and lung parenchyma 4, 23.
Inflammation in cystic fibrosis
Characteristics of airway inflammation
The most characteristic feature of airway inflammation in CF is a marked and persistent influx of polymorphonuclear neutrophils drawn into the airway lumen by host and bacterial chemoattractants. The chemoattractants in the CF lung that contribute to the chronic neutrophil influx include soluble bacterial products, fragments of the fifth component of complement, such as C5a and C5adesArg, and cellular products, such as interleukin (IL)-8 and leukotriene (LT) B4 3, 4, 23. IL-8, the major neutrophil chemoattractant peptide, is produced by a variety of cell types including neutrophils. Levels of IL-8 are increased in the sputum, bronchoalveolar lavage fluid (BALF), and serum of CF patients, even during stable clinical conditions 24, 25. These mediator levels correlate with the number of neutrophils in the BALF. Di Mango et al. 26 have shown that binding of P. aeruginosa to the aGM1 receptors, which are present in higher levels on CF airway epithelial cells, induces activation of nuclear factor (NF)-κB and hence, IL-8 expression. P. aeruginosa products also increase IL-8 production by airway epithelial cells 27. Thus, inflammatory cells and micro-organisms sustain IL-8 levels in CF airways and perpetuate a positive feedback loop that amplifies the inflammatory response. Moreover, recent studies suggest that CFTR dysfunction may be an initial cause of increased IL-8 levels in CF airways (see later). Increased concentrations of other pro-inflammatory cytokines, namely IL-1, tumour necrosis factor-α, and IL-6, have been detected in CF sputum and BALF, and are known to amplify airway inflammation 25, 28.
High levels of chemoattractants and pro-inflammatory cytokines ensure that activated neutrophils continue to accumulate in the airways, the decomposition of which is the major source of the deoxyribonucleic acid (DNA), which makes CF sputum so tenacious. Exacerbations of lung infection are accompanied by increased amounts of DNA in the sputum. This is primarily of human, rather than bacterial, origin 29. Neutrophils also release an array of potentially noxious mediators, such as reactive oxygen species (ROS) and proteolytic enzymes, which may damage the surrounding tissue (see later).
Onset and mechanisms of airway inflammation
The use of bronchoalveolar lavage (BAL) has allowed for major advances in understanding of the inflammatory process in CF lung. BAL studies in CF infants and patients with mild lung disease have provided insight into the time of onset and the relationship between airway infection and inflammation in CF lung disease. These studies have suggested that inflammation is a very early event and occurs in patients with mild disease as well as in stable clinical conditions 30–32. Konstan et al. 30 found that neutrophil-dominated airway inflammation is always present in CF patients >12 yrs of age and that even those with mild disease or remote from clinical exacerbations had large numbers of neutrophils and active elastase in their airways.
Khan et al. 31 and Armstrong et al. 32 have shown that airway inflammation is present in >40% of very young infants. This was identified by newborn screening, before lung disease became clinically apparent. Surprisingly, neutrophil-dominated inflammation and increased levels of IL-8 were present in the BALF of some infants who had no apparent signs of infection. Other studies have confirmed the presence of airway neutrophilia, despite negative cultures for the usual CF-associated pathogens, suggesting that inflammation may even precede infection 33. Moreover, Muhlebach et al. 34 recently reported that BALF from infected children with CF contained significantly more IL-8 and neutrophils than BALF from infected children with other respiratory conditions. In addition, in children with CF inflammatory response was higher than that in control subjects, even when bacterial quantity was taken into account 34. Thus, excessive inflammation in the CF lung may be related to constitutive abnormalities in the regulation of inflammatory response, independent of infectious stimuli. There is both in vivo and in vitro evidence for the argument that defective CFTR function may be directly related to heightened inflammation.
Increased numbers of inflammatory cells have been observed within the airway submucosa of CF mice raised in pathogen-free conditions, compared with wild-type litter-mates 35. Furthermore, it has been demonstrated both in vivo and in vitro that ΔF508 CF bronchial submucosal gland epithelial cells exhibit a selective upregulation in IL-8 production 36, associated with constitutively activated NF-κB and high IκB kinase α levels, observed at any extracellular hypo-, iso- and hypertonic NaCl content 37. The constitutive and/or excessive production of IL-8 by CF bronchial gland cells, which represent the predominant sites of CFTR expression in human airways, could implicate these cells in facilitating neutrophil recruitment associated with airway inflammation in CF patients. In contrast, it has been shown that CF bronchial epithelial cells produce decreased levels of the anti-inflammatory cytokine IL-10 compared to control cells. Lower levels of IL-10 have also been detected in BALF from adult CF patients 38. These observations further support the view that CFTR dysfunction is linked to a dysregulation of the inflammatory response, and that abnormal inflammatory control mechanisms favour a pro-inflammatory cytokine balance in the CF lung. There is also evidence that this dysfunction may be involved in the oxidant/antioxidant imbalance in CF airways (see later).
It has been reported that both myeloperoxidase-dependent oxidant activity and chloramine release are higher in CF patients than control subjects, and that both are inhibited by treatment of CF neutrophils with amiloride or choline buffer 39. These data suggest that intracellular pH or ion concentrations may be involved in the altered myeloperoxidase activity of CF neutrophils. Since CF gene expression has been reported in neutrophils 40 and CFTR has been implicated in the regulation of pH in intracellular organelles, the basic genetic defect may, therefore, contribute to excessive oxidant generation within CF airways. CFTR has also been implicated in the regulation of levels of glutathione 41, an important antioxidant of the respiratory tract, and its dysfunction may also account for the reduced levels reported in CF patients 42. Lastly, it has been suggested that CFTR mutations may lead to abnormal apoptosis in epithelial cells and leukocytes 43. This may potentially represent another mechanism for the heightened inflammatory response in CF. Several in vivo and in vitro findings thus support the argument that the basic genetic defect in CF contributes to marked and persistent inflammation within the airways. However, further studies are needed to firmly establish whether inflammation does precede infection and to define the exact sequence of events leading to CF lung disease.
Role of the inflammatory process in lung damage
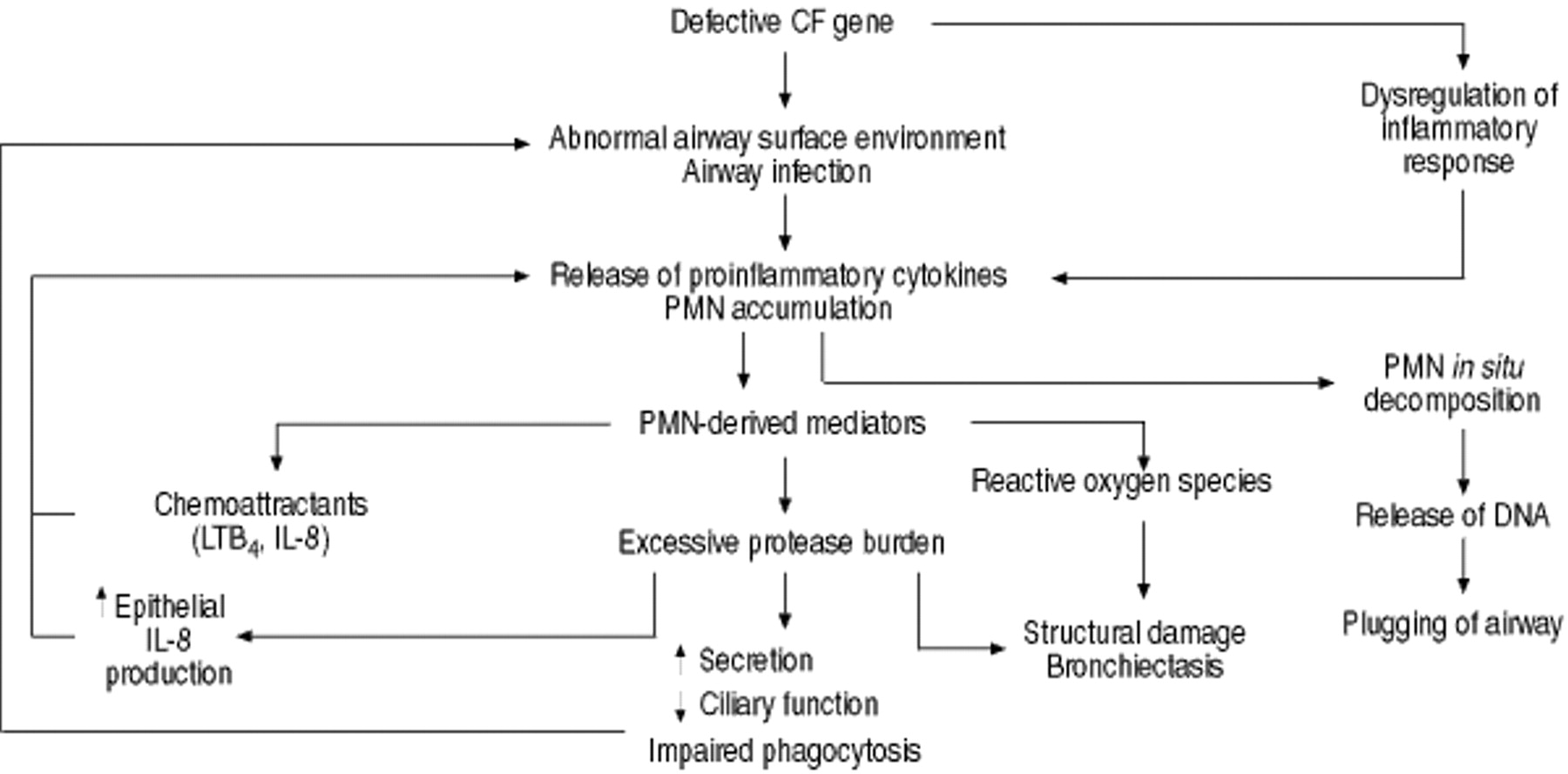
Regardless of how the inflammatory process is initiated and the temporal relationship between infection and inflammation in CF, there is little doubt that the persistent inflammatory response in the airways plays a central role in the progression of lung damage. Not only does the inflammatory response fail to eradicate the organism that incites inflammation, but accumulating evidence suggests that it actually contributes to local host defence impairment, which interferes with such eradication. Thus, a vicious circle of infection and inflammation is established, leading to structural damage of the lung tissue (fig. 1⇓).
Chronic airway infection and high levels of chemoattractants and inflammatory mediators ensure that activated neutrophils continue to accumulate in the airways. In addition to producing chemotactic factors and pro-inflammatory cytokines, both of which amplify and perpetuate inflammatory cell recruitment, these cells release an array of noxious mediators that may damage the surrounding tissue. Neutrophil elastase is one of these mediators and plays a major role in the pathophysiology of chronic inflammation in CF. It directly contributes to tissue damage by degrading structural proteins, such as elastin, collagen, and proteoglycans 44, and has many other detrimental biological activities in the CF airways. It is a potent secretagogue and enhances macromolecular secretion from serous gland cells 45. It also promotes hypertrophy and hyperplasia of the mucus-secreting apparatus and inhibits ciliary beating in vitro 46, 47. Taken together, these effects may further impair mucociliary clearance and exacerbate airway obstruction in CF patients. Neutrophil elastase also facilitates the persistence of infection by cleaving immunoglobulins, complement components and opsonic receptors, such as complement receptor (CR) 1, on the surface of phagocytes, and thus has an important impact on opsonophagocytosis 48–50. Furthermore, neutrophil elastase in secretions may itself attract more neutrophils into the airway lumen by inducing IL-8 production from epithelial cells 51.
The increased amount of free elastase in the airways of CF patients is related to both the large number of neutrophils and inactivation of the two main physiological inhibitors of proteases, α1-antitrypsin (α1-AT) and the secretory leukoprotease inhibitor. Two major mechanisms inactivate α1-AT in the CF lung: 1) cleavage of complexed inhibitor by excess, uncomplexed enzyme and/or Pseudomonas proteases not susceptible to its inhibitory activity; and 2) inactivation caused by ROS released during neutrophil activation 4, 23. Immunoblots of CF sputum show small amounts of α1-AT migrating as high molecular weight complexes with proteases, whereas the majority of the inhibitor is in the form of low molecular weight degradation products 52.
As a consequence of the excessively neutrophil-dominated inflammation, there is also a chronic burden of ROS in the respiratory tract of CF patients. Although the role of ROS as mediators of tissue damage in CF lung disease has not yet been clearly defined, abundant indirect evidence supports their involvement 53. High sputum levels of extracellular myeloperoxidase, a polymorphonuclear neutrophil (PMN)-derived enzyme, which transforms hydrogen peroxide into highly reactive oxygen metabolites, have been detected in CF patients and inversely correlated with lung function 54. Higher concentrations of oxidation products have also been reported in the plasma of CF patients compared with control subjects 55. Furthermore, levels of glutathione, the major local antioxidant in the lung, are reduced in both the epithelial lining fluid and the plasma of CF patients 42; a deficiency that predisposes patients with CF to oxidative tissue damage 42. Moreover, an increased propensity of eosinophils from CF patients to release cytotoxic proteins has recently been reported, suggesting that these cells and their mediators may contribute to progressive tissue damage in CF 56.
In summary, early in the course of CF lung disease, a self-perpetuating vicious circle of infection and inflammation is established. The excessive inflammatory response not only fails to clear infection, but contributes to its persistence and is mostly responsible for lung damage and the progression of CF lung disease.
Inflammatory response markers in cystic fibrosis
The recent acknowledgement that airway inflammation plays a crucial role in progressive lung damage in CF has encouraged the search for reliable markers of its severity. BAL, together with evaluation of neutrophil number and levels of inflammatory mediators, provides the most accurate picture of airway inflammation. However, routine bronchoscopy with BAL is unpleasant for the patient and is usually of no immediate benefit to the management of individual cases. Surrogate markers, collected by noninvasive procedures, would therefore be of great assistance.
Several markers have been evaluated in the sputum of CF patients and related to the severity of inflammation. It has been found that active-elastase sputum levels increase with increasing pulmonary involvement in clinically stable CF patients 57. In contrast, in a prospective study, Suter et al. 58 found a significant decrease in these levels after antimicrobial treatment. High levels of other neutrophil proteases, such as active collagenase, have been reported in CF sputum and are inversely related to disease severity 59. Other neutrophil mediators, such as oxidants (chloramine, taurine and myeloperoxidase) 54, chemoattractants (LTB4) 60 and several pro-inflammatory cytokines 25, have been detected in increased amounts in CF sputum. However, although sputum is easily collected, this can only be performed in individuals old enough to expectorate. Furthermore, sputum assays for some of these mediators are not reproducible.
Recently, it has been reported that nitrite levels are elevated in the breath condensate of CF patients compared to exhaled nitric oxide, which is normal or even low 61, 62. This is a potential marker in order to follow the inflammatory process that requires further long-term studies.
None of the numerous serum and plasma assays investigated as potential inflammatory markers have proved highly predictive of acute changes. C-reactive protein and the elastase-α1-antiprotease complex are frequently increased during exacerbations and fall after antibiotic therapy. However, a wide variability within and between patients has been observed in long-term studies 63. Two inflammatory markers in urine, LTE4 and desmosine (a break-down product of elastin), have been advocated as surrogate markers, but their variability is also high in long-term studies 64, 65. The evaluation of serum levels of soluble forms of leukocyte-endothelial adhesion molecules in clinically-stable CF patients has shown that soluble intracellular adhesion molecule (sICAM)-1 and soluble E-selectin (sE-selectin), but not soluble vascular cell adhesion molecules (sVCAM)-1 are significantly elevated compared with healthy controls. sICAM-1 levels can be correlated with disease severity. Some patients had serial measurements of soluble adhesion molecules when they were clinically stable and both before and after antibiotic treatment for a pulmonary exacerbation. Interestingly, serum levels of sICAM-1 and sE-selectin increased in all patients at the time of the exacerbation compared with levels at the time of stable clinical conditions, and were significantly reduced by a 2-week course of intravenous antibiotic treatment 66.
Increased levels of soluble adhesion molecules in serum and other body fluids have been demonstrated in several inflammatory diseases. It has been suggested that their measurement might be useful in monitoring inflammatory/immune disorders. The increase observed in the present CF patients could reflect an upregulation of these molecules on vascular endothelial cells, which may play an important role in the pathogenesis of CF lung inflammation, and could be an indication of its severity and persistence. Furthermore, the author's observations suggest that their measurement could assist in the follow-up of CF patients and in monitoring antibiotic therapy. Long-term studies are needed to substantiate the clinical potential of these markers.
Conclusions
A marked and persistent inflammatory process occurs very early in the course of CF lung disease and is present even in mild, stable disease. A growing body of evidence suggests that there is a link between CFTR dysfunction and the abnormal inflammatory response occurring in the airways.
Recognition that airway inflammation is primarily responsible for lung damage has provided a rationale for developing new therapeutic strategies for its control. While gene replacement therapy may eventually become the treatment of choice for patients with cystic fibrosis, at present, reducing the lung damage associated with airway inflammation is likely to have the greatest impact upon disease progression. A better understanding of the mechanisms of inflammation and the dynamics of the inflammatory response to infection in cystic fibrosis is essential. Furthermore, carefully-controlled studies are now needed to determine the best biological indicators of the activity of the inflammatory process and to define simple reliable markers for clinical follow-up of patients and monitoring the efficacy of therapy.

{kind=link}
The inflammatory response in the cystic fibrosis (CF) lung. The excessive inflammatory response in CF plays a major role in lung damage. A basic genetic defect leads to airway infection and dysregulation of inflammatory response, which both contribute to a persistent polymorphonuclear neutrophil (PMN) influx. In addition, chemoattractants released by PMNs themselves and interleukin (IL)-8 production induced by high levels of elastase in the airways maintain the chronic PMN recruitment. These cells release an array of noxious mediators, such as reactive oxygen species and proteolytic enzymes, which cause structural damage. The excessive protease burden also impairs phagocytosis, facilitating the persistence of infection. Thus, a self-perpetuating vicious circle of infection and inflammation is established, leading to irreversible lung damage. LTB4: leukotriene B4; DNA: deoxyribonucleic acid.
Footnotes
-
↵Previous articles in this Series: No. 1: Pilette C, Ouadrhiri Y, Godding V, Vaerman J-P, Sibille Y. Lung mucosal immunity: immunoglobulin-A revisited. Eur Respir J 2001; 18: 571–588. No. 2: Lambrecht BN, Prins J-B, Hoogsteden HC. Lung dendritic cells and host immunity to infection. Eur Respir J 2001; 18: 692–704. No. 3: Moore BB, Moore TA, Toews GB. Role of T- and B-lymphocytes in pulmonary host defences. Eur Respir J 2001; 18: 846–856. No. 4: Message SD, Johnston SL. The immunology of virus infection in asthma. Eur Respir J 2001; 18: 1013–1025. No. 5: Crameri R, Blaser K. Allergy and immunity to fungal infections and colonization. Eur Respir J 2002; 19: 151–157.
- Received March 22, 2001.
- Accepted May 8, 2001.
- © ERS Journals Ltd
References









