






Abstract
Toll-like receptors (TLRs) playan essential role inthe detection of invading pathogens and in theinduction of host antimicrobial defenses. TLR4, the major endotoxin receptor, and TLR2, with agonists derived principally from gram-positive organisms, are likely to be important in the pathogenesis of sepsis. Both TLR2 and TLR4 agonists regulate important neutrophil functions, including adhesion, generation of reactive oxygen species, and release of chemokines, and activate major proinflammatory signaling pathways, including the nuclear factor—κB path way. TLR stimulation produces on lya modest direct inhibition of neutrophil apoptosis, although this signal is great lyamplified by the presence of monocytes, suggesting that regulation of the lifespan of neutrophils by TLR agonists may beprincipally mediated by responses of other end otoxin-responsive cells. We suggest that activation of neutrophils by TLR sis highly regulated, permitting acute neutrophil antimicrobial responses to TLR activation while providing a “brake” on inflammation by requiring the presence of mononuclear cells to significantly extend neutronphil survival.
Neutrophils are, at least numerically, the major cell type participating in innate host responses to bacterial infection. They are central not only to the successful elimination of bacterial sepsis but also to sepsis-induced organ dysfunction. Our understanding of the innate immune response to gram-negative bacteria was transformed by the identification of a specific host membrane protein, CD14, that binds to endotoxin (lipopolysaccharide [LPS]) [1]. More recently, toll-like receptor4 (TLR4) was identified as a key signal-transducing molecule following LPS binding that initiates host mechanisms of antimicrobial defense [2, 3]. The TLR family currently includes 10 human TLR s that function as pattern recognition receptors for a broad range of predominantly microbial stimuli [4]. Neutrophils appear to express all of these receptors, except TLR3, at least at the mRNA level[5]. TLR4 is regarded as the major LPS receptor [2, 6], whereas cellular responses to components of gram-positive bacteria are mainly mediated via TLR2 [3]. TLR2 agonists include bacterial lipopeptides, lipoteichoic acids(LTAs), and peptidoglycans and signal via TLR2 heterodimers (TLR2 in combination with TLR1 or TLR6) [7–9].TLR2 and TLR4 are, thus, predicted to be of central importance in the pathogenesis of sepsis. Here, we discuss the roles of TLR2 andTLR4 in the regulation of neutrophil proinflammatory functions and apoptosis.
Effects Of TLR Agonists On Neutrophil Proinflammatory Functions
We have studied the roles of TLR2 and TLR4 in the regulation of neutrophil function (figure 1). Human peripheral blood neutrophils express both TLR2 and TLR4, albeit at lower levels than do monocytes [5, 10, 11], and also express CD14[12]. Expression of TLR2, but not TLR4, on the neutrophil surface is increased by pro inflammatory cytokines [11], and TLR2 is also up-regulated after exposure to a range of different bacteria [13].
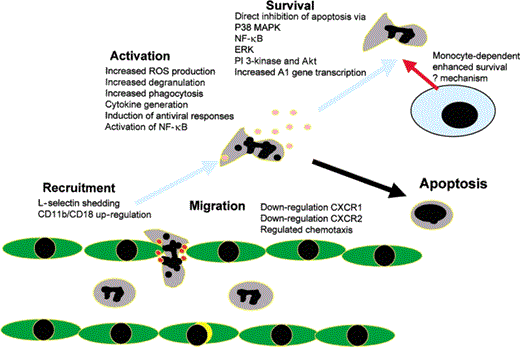
Roles of toll-like receptors (TLR s) in regulating neutrophil function. TLR2and TLR4 have been shown to modulate arange of neutrophil functions, as indicated. TLR-mediated extension of neutrophil survival ismediated via both directand indirect (monocyte-dependent) actionson the neutrophil. ERK, extra cellular signal—regulated kinase; MAPK, mitogen-activated protein kinase; NF, nuclear factor; PI 3–kinase,phosphoinositide 3–kinase; ROS, reactiveoxygen species.
Because commercially available LPS contains a significant amount of bacterial lipopeptide, it can be viewed as a dual TLR2 and TLR4 agonist [7]. We, therefore, used repurified LPS (pLPS), which lacks contaminating bacterial lipopeptides, as a TLR4 agonist [7]. A synthetic lipopeptide, Pam3CSK4, and peptidegly can purified fromStaphylococcus aure us were used as TLR2 agonists [7, 14].We examined the effects of these TLR agonists in functional assays reflecting crucial stages in neutronphilic inflammation: recruitment (assessed by adhesion molecule and chemo kinereceptor expression), activation (generation of reactive oxygen species [ROS] and production of IL-8), and life span (death by apoptosis). These experiments were done with highly purified neutrophils prepared by gradient centrifugation, followed by magnetic bead separation[5].
We found that activation of either TLR2 or TRL4 resulted in shedding of L-select in (CD62L) and up-regulation of CD11b at the neutrophil surface, in a concentration-dependent manner [14]. Other studies have also shown the ability of TLR agonists to induce shedding of L-select in [5, 15, 16]. We an dothers have found that both TLR2 and TLR4 agonists cause rapid down-regulation of CXCR2, the major neutrophil chemokine receptor, with a lesser effect on the more selective IL-8 receptor, CXCR1 [14] (authors' unpublished data). Hayashi et al. [5] found that CXCR1 and CXCR2 are down-regulated in response to TLR2 or TLR4 agonists, along with impaired chemotaxis to IL-8. In our studies, down-regulation of neutrophil chemo kine receptors did not result in a complete abolition of chemokine responses, as measured by under-agarose chemotaxis assays, suggesting a mechanism that could regulate, rather than abolish, responsiveness to chemo kine gradients in tissues and potentially regulate trafficking of neutrophils at sites of inflammation (authors' unpublished data). Thus, TLR agonists can potentially modulate neutrophil phenotype to regulate recruitment into tissues.
Both TLR2 and TLR4 agonists also induce neutrophil activation. In our studies, both caused a modest direct induction of the respiratory burst in highly purified neutrophil sand a more significant priming of the for my lmethionylleucyl phenylalanine-induced respiratory burst [14], in keeping with other data[5, 11, 15]. The dependence on TLR4 for generation of ROS by neutrophils has also been demonstrated in inflammatory peritoneal exudate neutrophils, with generation of ROS absent incells derived from C3H/HeJ mice that lack functional TLR4 [17]. Although TLR sare coupled directly in to activation of new gene transcription via the nuclear fact or (NF)–κB pathway [4],there is also evidence that, in neutrophils, this is amplified by generation of ROS [18]. Both TLR2 and TLR4 agonistscan induce IL-8 generation [14], again an effect observed in other studies[5, 11, 15]. This effect can be partially abrogated by a p38 mitogen—activated protein kinase (MAPK)inhibitor, SB203580 [14]. Although it is not possible to directly compare molarities of the different agonists used, agonists of both receptors do appear to be fully active in these functional assays. Recent work by Malcolm et al. [19] has demonstrated that LPS induces a wide range of transcriptional responses in neutrophils, up-regulating genes important for cell growth and survival and antiviral responses, in addition to the up-regulation of cytokine and chemokine genes. The latter, interestingly, include monocyte chemotactic protein (MCP)–1,suggesting that TLR-mediated effects on neutrophils may also include the ability to influence recruitment of other innate immune cell populations [19]. These LPS responses again show some dependence on p38 [20]. Expression of antiviral genes is induced by TLR4 but not TLR2 agonist [21].TLR2 ligands can, however, also induce cytokine and chemokine production, with involvement of phosphoinositide 3-kinase and the Akt signaling pathway[22].
Do Tlr Agonists Regulate Neutrophil Life Span And Death By Apoptosis?
Neutrophil apoptosis is widely accepted as a central mechanism in the resolution of inflammatory responses, leading to loss of proinflammatory functions of neutrophils and to specific recognition and clearance by professional phagocytes, such as tissue macrophages [23]. The constitutively shortlife span of then eutrophil can be extended by exposure to awide range of cytokines and proinflammatory mediators, and this mechanism is potentially important for extending the life span of neutron phils at the inflamed site and, thus, increasing the number of neutrophils available to participate in an immune response [24]. Dysregulation of neutrophil apoptosis by bacteria and their secretedpro ducts may be an important mechanism of disease, with premature apoptosis potentially resulting in failure of host defenses [25], where as a delay of neutrophil apoptosis could result in death of necrotic cells and, thus, in increased damage to host tissue [26]. TLR signaling has differing effects on apoptosisin different cell types. TLR4 can activate a Jun N-terminal kinase (JNK)/caspase—dependent pathway of apoptosis in endothelial cells [27], whereas, in monocytes, TLR2 activates both pro-survival and pro-death pathways, the latter via recruitment of Fas-associated death domain and downstream caspase activation [8]. However, a large number of studies show that LPS delays constitutive apoptosis of neutrophils [28, 29]. LPS priming can delay apoptosis of neutrophils in vivo and greatly augment host defenses in models of bacterialseps is [30]. Of interest, increased neutrophil apoptosis by influenza A virus in the presence of Streptococcus pneumoniaehas been postulated tobe a mechanism under lying increased risks of pneumonia after infection with influenza virus [31].
In studies using highly purified neutrophils, we demonstrated that TLR2 or TLR4 agonists, Pam3CSK4 or peptidogly can and pLPS, respectively, had only modest inhibitory effects on neutrophil apoptosis. pLPS, for example, produceda 50% inhibition of apoptosis after 4th in culture, when background apoptosis was negligible, but had no measurable effect on apoptosis at 22 h, when levels of constitutive apoptosis were high [14]. Extended time course experiments confirmed that a concentration of LPS that is effective in other functional assays caused only a very modest delay of constitutive apoptosis of neutrophils [14, 32]. However, the addition of unstimulated PBMCs to the cultures produced a delay of apoptosis equivalent to that caused by LPS, and, of importance, there was a synergistic effect of LPS and PBMCs to cause a very substantial delay of neutrophil apoptosis [32]. LPS therefore delay sneutrophil apoptosis via both direct and indirect actions.
Our data supported a role for NF-κB, and also MAPKs, including p38 and extra cellular signal—regulated kinase, indirect effects of pLPS on neutrophil apoptosis [14] and, thus, appeared to involve signaling pathways similar to those implicated in TLR activation of other proinflammatory neutrophil functions, as described above [14, 18, 22], and in there gulation of constitutive neutrophil apoptosis [33, 34]. All of these signaling pathways are recognized as potential therapeutic targets for treatment of neutrophilic inflammation in the context of acute lung injury [35].
The mechanism by which mononuclear cells cause delay of neutrophil apoptosis is unknown. Although IL-1β has been implicated as an autocrine orparacrine survival factor for LPS-stimulated neutrophils [36], we found that IL-1β has no direct effect on apoptosis of highly purified neutrophils and that the IL-1 receptor antagonist (IL-1RA) was unable to abrogate the antiapoptotic effects of either pLPS or PBMC. Moreover, in studies using neutrophils from IL-1R1–deficient mice, which are deficient in IL-1β signaling, LPS was still able to induce neutrophil survival (authors' unpublished data). Thus, effects of IL-1β on neutrophil survival must be indirect, occurring via effects on mononuclear cells. In contrast, however, we have recently examined the role of PBMCs in the initiation of neutrophil recruitment into tissues. Cocultures of primary airway smooth muscle cells show that they release IL-8 in response to LPS stimulation only in the presence of low numbers of PBMCs. This effect of PBMC s is IL-1β–dependent (authors' unpublished data). Interestingly, therefore, the factors responsible for mediating mononuclear cell effects on neutrophil survival and initiating production of neutrophil chemokines appear to be different, with the latter being IL-1β dependent. The nature of the mononuclear cell—derived antiapoptotic signal is a subject of ongoing investigation.
The potential importance of the relatively modest direct antiapoptotic action of TLR4 is that it suggests that neutrophil activation and, thus, immediate anti microbialactions may be at least partially separated from the extension of the life span of neutrophils. Such a system potentially offers a further regulatory step in the evolution of the innate immune response and could permit prompt resolution of a minor inflammatory reaction, with out the creation of a pool of highly activated neutrophils with an extended life span and, thus, an increased capacity for injury to host tissue. Because TLR agonists caninduce neutrophil survival viaactions on mononuclear cells, this implies the existence of a role for the monocyte/macrophage in gradation of the neutrophilic inflammatory response and, also, that differences between pathogens in the relative recruitment of neutrophils and monocytes might, via TLR responses, alter the duration and extent of the host immune eresponse.
Unanswered Questions
There are a number of major areas in which our understanding of neutrophil responses to TLR2 and TLR4 are incomplete.
What is the composition of TLR signaling complexes on the neutrophil cell surface, and how is TLR signaling modified by engagement of multiple receptors simultaneously? Many of the studies referenced above have examined the role of small-molecule TLR agonists in inducing cellular responses. Innate immune cells have developed to respond to whole pathogens, and bacterial cell wall components also typically aggregate, so that the physiological interaction between pathogen and cell occurs over a large surface area and involves presentation of a range of bacterial components. LPS responses are partially dependent on activation of coreceptors, such as CD11b/CD18 [37], and it is unclear whether LPS is most active in multimeric forms[38] or as monomers complexed with myeloid differentiation protein—2 [39]. In addition, the cellular response may result from integration of signals received from multiple pattern recognition receptors and other proteins, particularly when considering whole or fragmented bacteria, rather than purified agonists, with receptor proteins perhaps aggregated within lipid rafts [4, 40, 41]. Of the accessory molecule simplicated in TLR4 signaling, neutrophils certainly express CD14, which is involved in the presentation of LPS to TLR4 [12], and CD11b/CD18, which is involvedin LPS signaling. Neutrophils also express other coreceptors implicated in responses tozymosan, including dectin-1 [42], but the expression and roles of other potential coreceptors have not been studied in neutrophils [4].
How are ligand-specific responses achieved? TLR2 can form heterodimers with either TLR1 or TLR6, broadening the range of stimuli (predominantly from gram-positive organisms) signaling via TLR2. These heterodimers may signal differently, which thus explains differences between, for example, Pam3CSK4 and LTAs in the magnitude of their antiapoptotic effects [14, 15]. Given the complex nature and composition of the TLR signaling platforms, it also seems probable that individual TLRs can generate specific responses to different pathogens by recruitment of different combinations of coerceptors.
What are the differences in effect or function in response to TLR2 or TLR4 signaling? At present, the evidence suggests that TLR2 and TLR4 have both shared and distinct actions on neutrophil signal transduction, with activation of a range of proinflammatory signaling pathways via adapter proteins [4]. TLR2 and TLR4 mediate share dactions, presumably via the adapters MyD88 and Mal, but TLR4 also activatesa panel of antiviral genes. Interestingly, activation of these genes is significantly dependent on type 1IFN generation in monocytes (the most studied cell with respect to TLR signal transduction [43]), but, in neutrophils, this response is IFN Independent [21]. Cell type—specific regulation of signaling pathways might underpin the preferential inhibition of, for example, TLR4 antiapoptotic pathways while preserving immediate antimicrobial responses. Clarification of these differences will be important, but the results will need to beviewed mindful of the physiological situation in which a single bacterial species can activate multiple TLRs (for example, S. pneumoniae canactivate TLR2 via LTA [44] and TLR4 via pneumolys in [45], as well as TLR9 via its DNA [4].
Potential Roles Of Tlr2 and Tlr4 In Treatment Of and Susceptibility To Sepsis
The possibility that down-regulation of TLR responsesis a therapeutic target for sepsis was first raised by the demonstration that mice with a spontaneous mutation of TLR4 are resistant to the effects of systemic endotox in [2, 6]. LPS or lipid A from Rhodobacter sphaeroides can prevent activation of human TLR4 by LPS[46], as does asynthetic lipodisaccharide, E5564 [47]. An antagonistic antibody against the extra cellular domain of TLR2 protected mice both against infusion of lipopeptide and against otherwise lethal challenge with Bacillus subtil is [48]. In addition, inhibition of TLR-dependent proinflammatory signaling pathways by specific products of microorganisms (e.g., vaccinia virus proteins that specifically inhibit interactions of IL-1 receptor—associated kinases/TNF receptor—associated factor 6)may identify novel strategies to inhibit signaling induced by bacterial pathogens [49]. Although a potential concern is whether such strategies would be of benefit in established sepsis and ameliorate host tissue injury, the association of genetically impaired TLR4 function with more chronic inflammatory conditions, such as atherosclerosis [50] and inflammatory bowel disease[51, 52], might suggest possible therapeutic benefit. Further understanding of TLR actions may also indirectly benefit patients who are at risk of sepsis, because TLR activators are extremely potent adjuvants that maybe useful for vaccine design [4].
A number of polymorphisms of TLR4 have been identified in the human genome. One relatively common polymorphism, Asp299Gly, maybe associated with an increased risk of septicshock [53]. The same polymorphism confers protection against atherosclerosis [50] but increased susceptibility to inflammatory bowel disease [52]. The Asp299 Glypolymorphism is not associated with increased risk of meningococcal disease [54, 55], although rarer TLR4 alleles may increase risk of this infection [55]. Other TLR4 polymorphisms have been associated with hyporesponsiveness to LPS [56]. Polymorphisms in TLR2 are also described [57]. Such studies havethe potential to aid the future identification ofpatients at risk of susceptibility to infection and thus of septic shock by combining genetic analysis with the identification of relevant patient phenotypes.
Conclusions
There is now evidence that TLR s, particularly TLR2 and TLR4,directly regulate major proinflammatory and host defense functions of human neutrophils. TLR responses are, however, tightly regulated, and cross-talk between TLR stimulated immune cells provides further modulation of the host immune response.
Acknowledgements
Financial support. Medical Research Council (Clinician Scientist Fellowship G108/388 to I.S.).
Potential conflicts of interest. I.S.,S.K.D., M.K.B.W.: no conflicts.


{kind=link}
Comments