Article Text

Statistics from Altmetric.com
Pulmonary arterial hypertension (PAH) is a progressive, life-threatening disease characterised by intense pulmonary vascular remodelling leading to high pulmonary arterial pressure, right ventricular failure and death. Although recent advances in therapies have proven to be effective in alleviating disease symptoms and improving functional capacity and survival, patients with PAH continue to suffer from persistent dyspnoea and significant exercise intolerance, which negatively impact their quality of life. In recent years, it has been increasingly recognised that exercise limitation in PAH is not merely due to right heart dysfunction and respiratory impairment, but is also a consequence of skeletal muscle abnormalities.1–3 Impaired skeletal muscle function, including reduced volitional and nonvolitional muscle strength and endurance2 4 5 decreased contractility,6 reduced capillary density and impaired oxygenation at the microcirculation level,4 7 as well as a shift towards type 2 muscle fibres,1 3 5 have been repeatedly documented in human PAH. Skeletal muscle atrophy has been less frequently reported3 and underlying mechanisms are not well studied.
To date, numerous metabolic and signalling abnormalities associated with muscle dysfunction in PAH have been uncovered. These include a shift from oxidative to glycolytic metabolism, suppression of signalling pathways responsible for a hypertrophic response (eg, Akt and S6K), elevation of negative regulators of muscle homeostasis (eg, myostatin and activin A) and engagement of ubiquitin–proteasome-mediated muscle proteolysis signalling (eg, atrogin-1 and MuRF1).3 8 9 Loss of skeletal muscle microcirculation mediated by microRNA-126 downregulation, as well as sirtuin-3/AMP-activated protein kinase inactivation, and cytokines (ie, tumour necrosis factor-α and interleukin-6)-regulated skeletal muscle insulin resistance and abnormalities of mitochondrial biogenesis have also been documented.4 8 10 The molecular mechanisms of skeletal muscle atrophy in PAH, however, are not well understood, and available human PAH-related data are limited by a small number of studies with only a few patients examined.
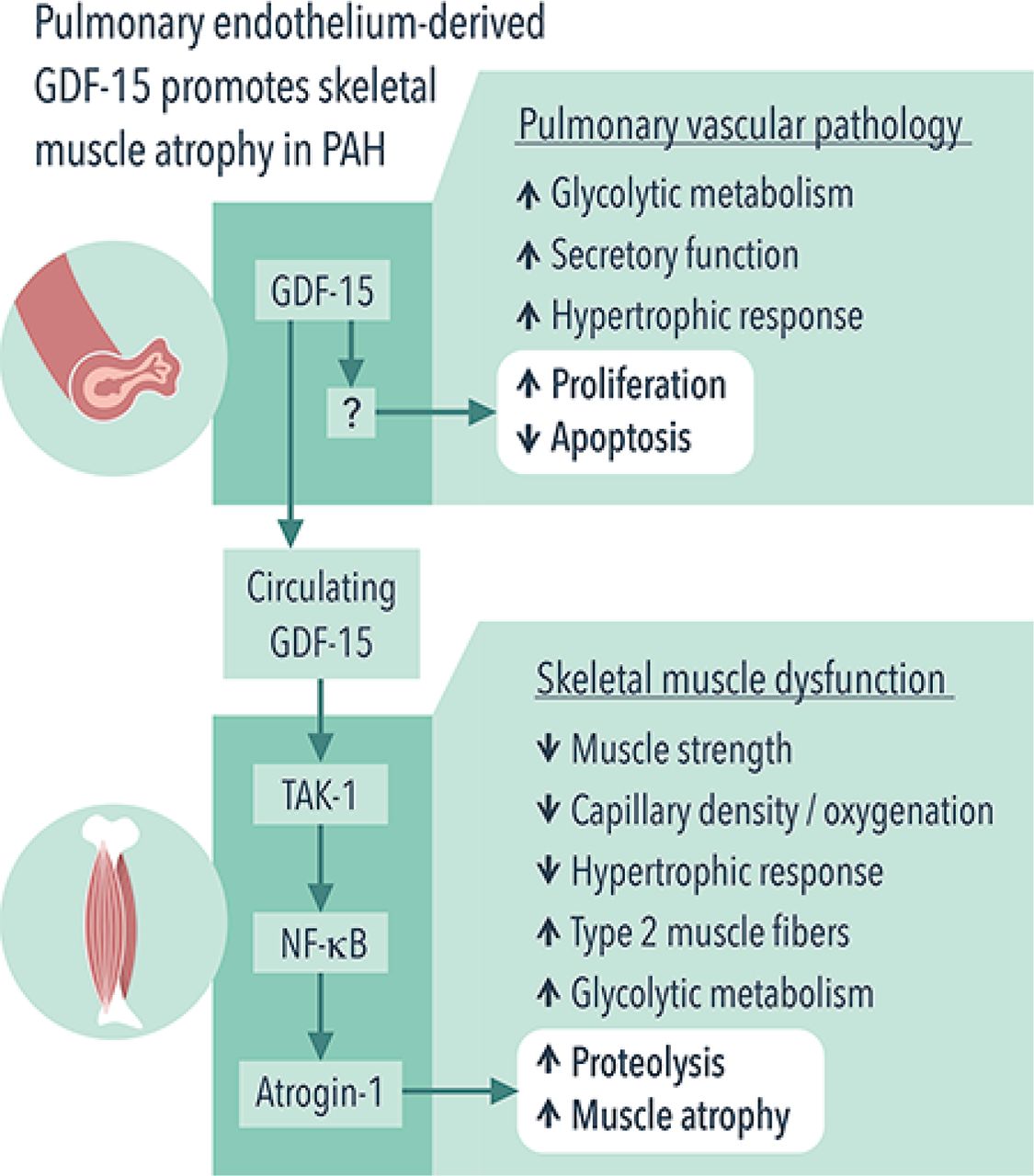
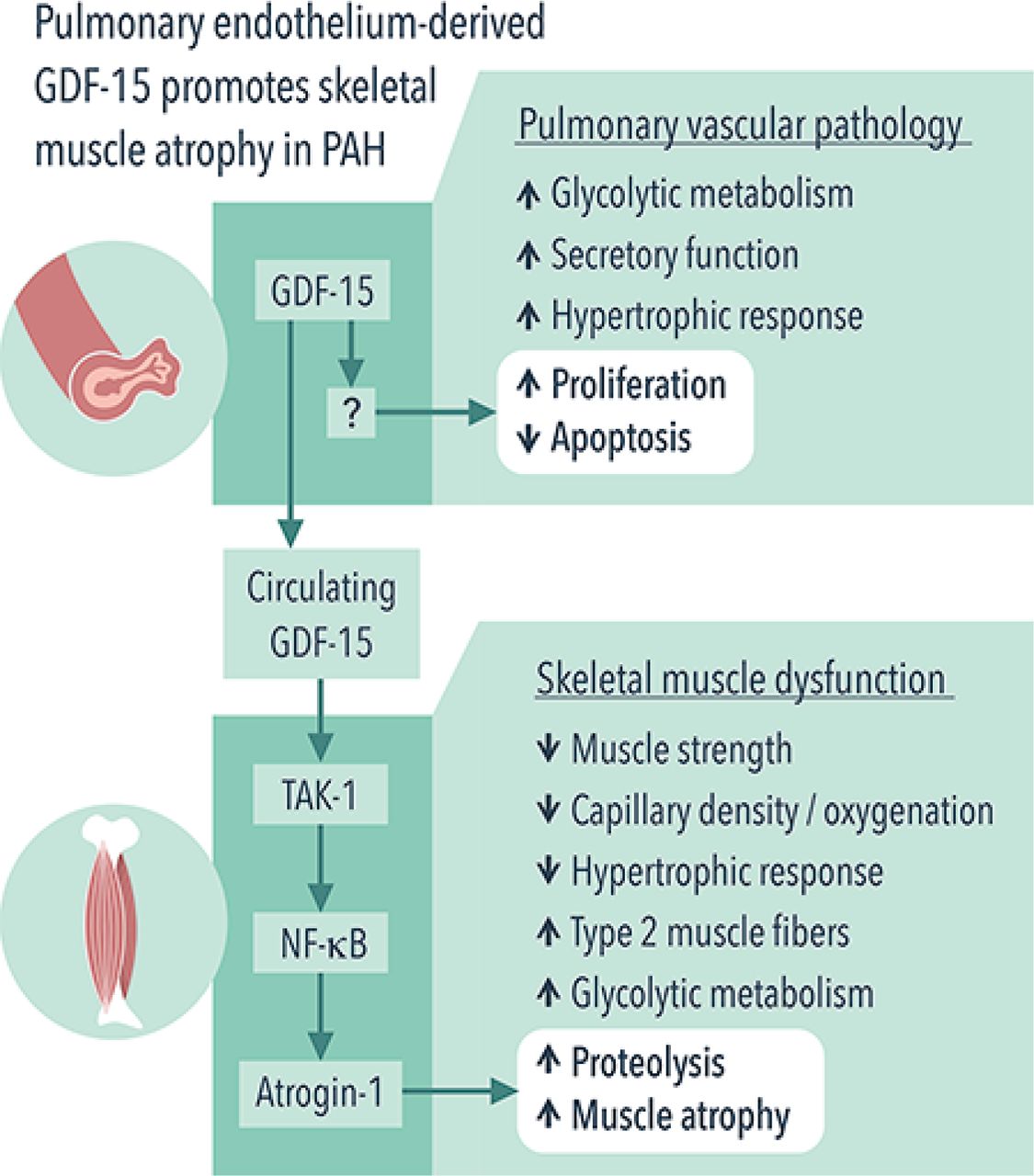
In this issue of Thorax, Garfield and colleagues reveal new role for growth differentiation factor 15 (GDF-15)/transforming growth factor β (TGF-β) activated kinase 1 (TAK-1) signalling as potential regulators of skeletal muscle atrophy in PAH11 (figure 1). Consistent with previous studies,12 13 the authors found that GDF-15, a member of the TGF-β/bone morphogenetic protein superfamily, is overexpressed in pulmonary vascular endothelial cells in rat monocrotaline (MCT) and mouse SU5416/hypoxia models of pulmonary hypertension (PH). The authors further demonstrate that this increase was associated with elevation of circulating GDF-15 levels, which, most importantly, correlated with a reduced diameter of tibialis anterior (TA) muscle fibre in both studied models, whereas it slightly correlated with quadriceps volume and function in human PAH. In addition, the association of GDF-15 and muscle atrophy was found in in vitro skeletal muscle cell culture. In aggregate, these data suggest that pulmonary circulation directly influences muscle mass in PAH through a GDF-15-mediated endocrine mechanism. Although provocative, this finding is not surprising since PAH is increasingly recognised as a systemic disease with metabolic, inflammatory, genetic and epigenetic abnormalities far beyond the pulmonary circulation.
{kind=link}
Pulmonary endothelium-derived GDF-15 promotes skeletal muscle atrophy in PAH. PAH manifests by both pulmonary vascular remodelling and skeletal muscle dysfunction. Garfield and colleagues11 report that, in PAH, pulmonary vasculature promotes skeletal muscle atrophy via endocrine mechanism. Specifically, pulmonary endothelium-derived GDF-15 activates TAK-1, which, in turn, upregulates atrogin-1 and induces muscle atrophy via NF-κB. GDF-15, growth differentiation factor 15; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PAH, pulmonary arterial hypertension; TAK-1, transforming growth factor β activated kinase 1;
The mechanisms by which GDF-15 promotes skeletal muscle mass loss remain, however, elusive. GDF-15 promotes weight loss and reduces muscle mass by suppressing appetite through interacting with glial cell-derived neurotrophic factor receptor α-like (GFRAL)14–16 and expedites muscle protein degradation via increasing expression of the ubiquitin ligases atrogin-1 and MuRF1 in C2C12 myoblasts.17 GDF-15 can also signal through TAK-1 and nuclear factor kappa light-chain-enhancer of activated B cells (NF-κB), as well as via their short-loop feedback system, in infection-associated epithelial stress and early tumour development.18 In this new study, TAK-1–NF-κB was found to mediate a direct muscle atrophic effect of GDF-15 through upregulating atrogin-1, a mechanism independent of appetite suppression, as GFRAL expression was not detected in skeletal muscle.14 The authors also found that targeting TAK-1–NF-κB with TAK-1 inhibitor 5(Z)−7-oxozeanol reduced GDF-15-mediated muscle atrophy in C2C12 myotubes and in rats with MCT-induced PH without significantly affecting circulating GDF-15 levels and pulmonary pressures. Overall, these findings suggest that GDF-15 and its downstream TAK-1–NF-κB signalling may be potential targets for future therapeutic intervention aimed at improving muscle mass, exercise tolerance and quality of life in patients with PAH.
The study by Garfield and colleagues is clearly advancing the field. There are, however, still some important questions that remain to be answered. First, the mechanisms of GDF-15 regulation and function in pulmonary vascular endothelium and plexiform lesions in the setting of PAH remain to be determined. To note, GDF-15 induces growth of cancer cells and prevents endothelial cell apoptosis via activating Akt,19 20 an important regulator of cell hyperproliferation and survival in PAH,21 suggesting a potential link between GDF-15 and Akt in pulmonary vascular remodelling and overall PAH. Additionally, the effects of circulating GDF-15 on cardiac musculature with a specific focus on right ventricle should be determined.
It is also uncertain whether the increased skeletal muscle loss is a secondary effect of GDF-15 released from the pulmonary vasculature and/or other organs or a local effect of GDF-15 in skeletal muscle. A better understating of these phenomena can help in optimising clinical decision-making and improving outcomes by different treatment strategies. It is also not clear whether patients with PAH enrolled in this study had significant skeletal muscle atrophy, as there was no comparison with age-matched and sex-matched control subjects. This point is critical since numerous studies failed to document muscle1 or muscle fibre1 4 6 7 atrophy in human PAH. Although this is likely explained by selection bias (most studies predominantly recruiting patients with WHO class II PAH, as in the current study), the evaluation of pathways implicated in this process may be less relevant in a study population without atrophy. It is also not clear whether patients with PAH enrolled in this study had any background therapies that may affect circulating levels of GDF-15, dependent or independent of improvement of pulmonary vascular endothelium. Therefore, the results-driven generalisations of this study requires further investigation.
Another limitation is the difficulty in extrapolating muscle-specific findings to other muscles. For example, tibialis anterior is phenotypically different from soleus or quadriceps. This is also exemplified in the current manuscript where GDF-15-mediated abnormalities were not observed in rats’ soleus. Even within a given muscle, fibre type distribution and myocyte characteristics differ according to their location within the muscle.22 Extrapolating findings from mouse myoblasts (C2C12 cell lines) and rodent PAH models carries limitations, especially in the absence of confirmation from human biopsies. It is also should be noted that, as presented, most correlations between circulating GDF-15 levels and muscle abnormalities are loose despite the fact that both PH and control rats were pooled together, and GDF-15 expression/mRNA levels were unchanged within PAH skeletal muscles. This suggests that there may be a limited cause–effect relationship, or indirect mechanisms are involved. Similarly, this association is confounded by the fact that GDF-15 is a biomarker of PH severity (which was very mild and highly variable in their ‘observation study’ with many MCT rats having no elevated pulmonary pressures, as expected with a low dose of MCT). Alternative experimental PAH model such as SU5416/hypoxia rats, which develops more severe plexiform lesions than MCT rats and SU5416/hypoxia mice, as well as endothelial cell-specific GDF-15 knockout animals, may serve as important tools to address the knowledge gap in the future.
Finally, similar to group 1 PAH, reduced skeletal and respiratory muscle mass and strength, a shift towards type 1 muscle fibres, reduced skeletal muscle aerobic enzyme activity and mitochondrial volume density have also been documented in other chronic cardiorespiratory diseases like heart failure with reduced or preserved ejection fraction and chronic obstructive pulmonary disease. Similarities in skeletal and respiratory myopathy observed within these conditions would suggest that a general underlying mechanism might be at play. While GDF-15 has been associated with muscle wasting in COPD, intensive care acquired weakness and cancer,17 23 24 it is more commonly described as a biomarker of disease severity predicting outcomes in these diseases as in acute pulmonary embolism, diabetes and renal failure. Therefore, the precise and independent contribution of GDF-15 in PAH-mediated muscle wasting and to which extent these abnormalities are specific to PAH remain unknown.
Garfield and colleagues should be commended for shedding new light on the relationship between GDF-15 and skeletal muscle atrophy in PAH, which paves the way for future studies. Future investigations should aim to test whether tackling GDF-15 can lead to improvement of skeletal muscle function, exercise tolerance and quality of life in PAH. Apart from providing new insight into the mechanism of muscle wasting in PAH through a lung–skeletal muscle crosstalk mediated by GDF-15, Garfield and colleagues also identify TAK-1–NF-κB signalling as potential targets for future therapeutic intervention to minimise muscle loss and to improve exercise limitation in PAH. An understanding that, in addition to playing the role in skeletal muscle atrophy, GDF-15 acts as a proproliferative/antiapoptotic agent for pulmonary vascular endothelial cells opens up a potential opportunity for developing therapeutics (ie, specific antibody) to target circulating GDF-15 in PAH. Nevertheless, there is still a long way to go before GDF-15 can be employed as a biomarker to predict muscle atrophy in PAH and to address the clinical relevance of GDF-15- and TAK-1-targeting strategies in patients with PAH.
References
Footnotes
Contributors All three authors had participated in drafting the work or revising it critically for important intellectual content. All three authors provided final approval of the version submitted.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned; internally peer reviewed.