Article Text

Abstract
Rationale Hypercapnia is common in mechanically ventilated patients. Experimentally, ‘therapeutic hypercapnia’ can protect, but it can also cause harm, depending on the mechanism of injury. Hypercapnia suppresses multiple signalling pathways. Previous investigations have examined mechanisms that were known a priori, but only a limited number of pathways, each suppressed by CO2, have been reported.
Objective Because of the complexity and interdependence of processes in acute lung injury, this study sought to fill in knowledge gaps using an unbiased screen, aiming to identify a specifically upregulated pathway.
Methods and results Using genome-wide gene expression analysis in a mouse model of ventilator-induced lung injury, we discovered a previously unsuspected mechanism by which CO2 can protect against injury: induction of the transporter protein for α-tocopherol, α-tocopherol transfer protein (αTTP). Pulmonary αTTP was induced by inspired CO2 in two in vivo murine models of ventilator-induced lung injury; the level of αTTP expression correlated with degree of lung protection; and, absence of the αTTP gene significantly reduced the protective effects of CO2. α-Tocopherol is a potent antioxidant and hypercapnia increased lung α-tocopherol in wild-type mice, but this did not alter superoxide generation or expression of NRF2-dependent antioxidant response genes in wild-type or in αTTP−/− mice. In concordance with a regulatory role for α-tocopherol in lipid mediator synthesis, hypercapnia attenuated 5-lipoxygenase activity and this was dependent on the presence of αTTP.
Conclusions Inspired CO2 upregulates αTTP which increases lung α-tocopherol levels and inhibits synthesis of a pathogenic chemoattractant.
- ARDS
- Assisted Ventilation
- Lung Physiology
- Respiratory Measurement
Statistics from Altmetric.com
Key messages
What is the key question?
Can molecular mechanisms induced by hypercapnia during mechanical ventilation reveal potential therapeutic pathways for lung injury?
Does hypercapnia protect against lung injury by activating protective genes?
What is the bottom line?
Inspired CO2 upregulates a transport protein (α-tocopherol transfer protein); this mobilises vitamin E in the lung which inhibits synthesis of a pathogenic chemoattractant, leukotriene B4.
Why read on?
Experimental elevation of CO2 can protect (or cause harm); this new induced mechanism of protection from CO2 provides new potentially testable therapies for lung injury.
Introduction
Hypercapnia is common in mechanically ventilated patients with injured lungs, where tidal volume is deliberately lowered to lessen the risk of ventilator-induced lung injury (VILI).1 However, accumulating evidence indicates that CO2 can have potent effects in injured tissue that are independent of changes in mechanical ventilation.2 Depending on the context, these effects may be protective, for example, attenuating reperfusion injury,3 acute sepsis4 or ventilator-induced injury;5 ,6 or, injurious, for example, impairing alveolar fluid clearance.7
Several molecular mechanisms of hypercapnic action have been described. For example, elevated CO2 inhibits expression of inflammatory cytokines,5 activation of nuclear factor κB (NFκB)8 and sheddase (a disintegrin and metalloprotease 17; ADAM17)6 signalling, and functional protein expression (eg, endocytosis of ion channels).7 However, these observations are limited in scope because all were based on injury processes known a priori. Also, many of the mechanisms are pivotal biological processes and thus have limited translational potential (eg, inhibition of NFκB).
We sought to address this knowledge gap using an unbiased screen. We chose a microarray approach because the technology is mature, and because regulation at the level of gene expression would be a proximal and fundamental step in most identified signalling or synthetic processes. Microarray analyses have led to identification of key molecular pathways in VILI9 and of genes regulated by hypercapnia in neonatal lung development,10 and provide the possibility of identifying a specifically upregulated mechanistic pathway. Hypercapnia-dependent transcriptional responses have been recently reviewed.11 While not fully characterised, they include inhibition of NFκB12 and hypoxia-inducible factor 1α (HIF-1α)13 responses, and activation of CREB14 and FoxO3a.15
We utilised a mouse model of VILI in which addition of CO2 to the inspired gas protects against injury.5 We found that hypercapnia activated few genes; however, hypercapnia was protective in VILI, and here it increased the expression of α-tocopherol transfer protein (αTTP). We describe how increased expression of αTTP may contribute to the protection afforded by hypercapnia in this multifaceted in vivo model.
Methods
Additional details can be found in the online supplementary data.
supplementary data
Animal model
All animal procedures were approved by the animal care committee of the Hospital for Sick Children (Toronto, Ontario, Canada) in accordance with the Guidelines of the Canadian Council on Animal Care. C57BL/6J male mice (20–25 g, Charles River, St Constant, Quebec, Canada) were anesthetised and ventilated: tidal volume (VT) 10 mL/kg, positive end-expiratory pressure (PEEP) 2.0 cm H2O, frequency 135/min, FiO2 0.21. Two models were employed. Model-1: mice were randomised to Severe Injury as previously described5 (peak inspiratory pressure 27 cm H2O, VT 35–40 mL/kg, PEEP 0 cm H2O, frequency 30–35/min) versus continuation of baseline (low VT) ventilation with normocapnia (FiO2 0.75, FiCO2 0, balance N2) or hypercapnia (FiO2 0.75, FiCO2 0.12, balance N2) for 3 hours. Model-2: mice were subjected to Moderate Injury (VT 20 mL/kg, PEEP 0 cm H2O, frequency 45/min) for 4 hours, randomised to normocapnia (room air) or hypercapnia (FiCO2 0.12, balance room air).
Genetically modified mice
Male knockout (αTTP−/−) (strain B6.129S4-Ttpa1Far/J, Jackson Laboratories, Sacramento, California, USA) and wild-type sibling (αTTP+/+) controls were subjected to severe injury (Model-1) and randomised to normocapnia or hypercapnia.
Microarray analysis
We performed gene expression analysis using lung samples from mice included in our previous study,6 ventilated with the Severe Injury protocol (Model-1). RNA from five groups: (1) non-ventilated, (2) low VT normocapnia, (3) low VT hypercapnia, (4) high VT normocapnia, and (5) high VT hypercapnia (n=5/group non-ventilated and low VT; n=10/group high VT) was hybridised to Affymetrix (Santa Clara, California, USA) mouse gene 1.0 ST arrays. Primary datasets are accessible through NCBI GEO (series accession number GSE86229). Data were subjected to robust multichip analysis normalisation; preliminary principal components analysis (PCA) identified two outliers which were subsequently excluded from downstream analysis. Analysis of changes in gene expression was performed using Partek Genomics Suite (Partek Inc, St Louis, Michigan, USA), by two-way analysis of variance (ANOVA) using ventilation (non-ventilated, high VT, low VT) and CO2 (normocapnia, hypercapnia) as factors, Benjamini and Hochberg false discovery rate 5%, and fold-change cutoff at 1.5. Overrepresented canonical pathways associated with VT and hypercapnia were identified using Ingenuity Pathway Analysis (IPA, Ingenuity Systems, Redwood City, California, USA).
RT-PCR
Changes in gene expression were measured using relative quantitative real-time PCR by the ddCt method. Primers are shown in online supplementary table S1.
supplementary tables
Inflammatory mediators
Myeloperoxidase (MPO) was measured in lung homogenates using o-dianisidine dihydrochloride and H2O2 as substrates. Cytokines in bronchoalveolar (BAL) fluid were quantitated using a Milliplex mouse Cytokine Immunoassay Kit (Millipore, Billerica, Massachusetts, USA). Eicosanoids in BAL were quantitated by liquid chromatography (LC)–tandem mass spectrometry (MS), with cysteinyl leukotrienes (CysLTs) measured by enzyme immunoassay (Enzo Life Sciences, Farmingdale, New York, USA). Superoxide radical in hydroethidine-treated mouse lung was detected through quantification of 2-hydroxyethidium using LC-MS-MS analysis.
Lung tissue α-tocopherol
Lung tissue α-tocopherol was quantitated using HPLC from homogenates spiked with internal standard δ-tocopherol (Sigma-Aldrich Canada Co., Oakville, Ontario, Canada).
Statistics
Data are presented as dot plots with mean (for normally distributed data) or median (non-normal distributions) indicated by a horizontal bar. Statistical analyses were calculated with Sigmaplot V.12.3 (Systat software Inc) using ANOVA for multi-group comparisons. Two group comparisons used t tests on normally distributed data, and Mann–Whitney U for non-normally distributed data. p values <0.05 were considered significant.
Results
Gene expression and hypercapnic protection against ventilator-induced lung injury
High VT ventilation was used to generate VILI in C57BL/6J mice, as previously published;6 CO2 (hypercapnia) was added to the inspired gas to protect against VILI in two of five experimental groups: (1) non-ventilated, (2) low VT normocapnia, (3) low VT hypercapnia, (4) high VT normocapnia, and (5) high VT hypercapnia.6 Lung injury was significantly greater in high VT normocapnia compared with all other groups, and hypercapnia protected against VILI.6 PaO2, PaCO2 and pH values are given in online supplementary table S2.
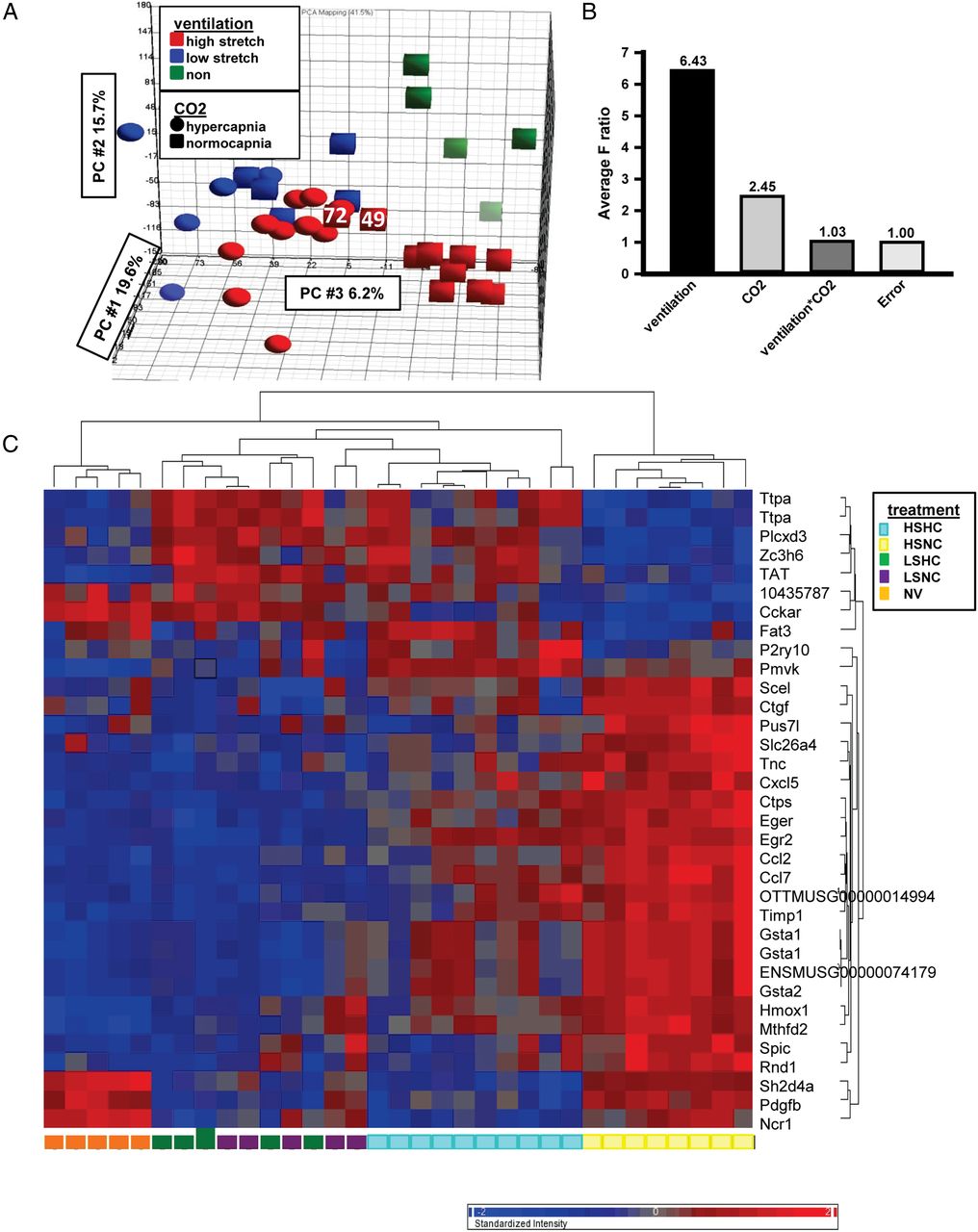
Microarray analysis of lung samples was used to determine gene expression patterns associated with hypercapnic protection. PCA confirmed that whole gene expression profiles were similar among samples within each group (figure 1A). Two-way ANOVA analysis of overall gene expression with ventilation and CO2 as factors both showed significant sources of variation, but no significant interaction (figure 1B). High VT ventilation (normocapnia) changed the expression of 1658 gene probe sets (contrasted with no ventilation or low VT; fold change 1.5, p<0.05), including large increases in inflammatory gene expression (see online supplementary table S3).

Microarray data in murine ventilator-induced lung injury and hypercapnia. (A) Principal component analysis plot for microarray data showing effects of ventilation and hypercapnia. Note: samples 72 and 49, from the high stretch normocapnia group, fell closer to the high stretch hypercapnia cluster than the high stretch normocapnia cluster and were excluded from further analysis. (B) Sources of variation in total gene expression estimated by two-way ANOVA. F ratio (y-axis) for each factor (x-axis) represents the F-statistics for that factor relative to F-statistic for error (noise) for all genes. (C) Cluster analysis showing changes in mRNA abundance during mouse lung ventilation as determined by microarray analysis. Each column represents RNA from a single mouse, and the heatmap shows genes detected by Robust Multichip Average analysis with expression increased (red) or decreased (blue) by 1.5 fold. HS, high stretch; LS, low stretch; HC, hypercapnia; NC, normocapnia; NV, non-ventilated.
In high VT ventilation, there were modest changes in gene expression when contrasting normocapnia with hypercapnia. Hierarchical clustering of this gene set (figure 1C) confirmed distinct expression patterns, with some overlap in low VT (uninjured) ventilation. A small number of genes were overexpressed in high VT hypercapnia versus high VT normocapnia, and most differentially expressed genes displayed downregulation under hypercapnia. Twenty-nine independent known genes responding to hypercapnia during high VT ventilation were identified (table 1). We selected 12 candidate genes from this list based on plausibly protective effect and regulation at the transcriptional level, and real time RT-PCR confirmed that nine were regulated by hypercapnia (gene symbols: Ttpa, Pmvk, Pdgfb, Ereg, Egr2, Tnc, Gsta1, Ncr1 and Hmox1); three trended as in microarrays, but were not significant (P2ry10, Cckar, Ctgf; table 1).
List of genes differentially expressed by hypercapnia in severe injury VILI
Identification of candidate gene αTTP in VILI
The most highly upregulated gene, encoding αTTP, was significantly more expressed in high VT hypercapnia versus high VT normocapnia (confirmed by RT-PCR; table 1). In addition, the degree of protection (eg, decrement in static compliance) from hypercapnia was proportional to the degree of expression of the αTTP mRNA (figure 2). We used a second model of more moderate ventilation injury (VT=20 mL/kg; FiO2=0.21; see online supplementary figure S1), in which hypercapnia attenuated injury (ie, BAL protein, lung MPO), and induced αTTP mRNA (figure 3).

Hypercapnic protection and expression of α-tocopherol transferase protein (αTTP). Linear regression analysis indicating correlation between αTTP mRNA expression (RT-PCR) and decrease in static compliance in ventilated mice. Dotted lines indicate 95% CIs. R=−0.73, p<0.001.
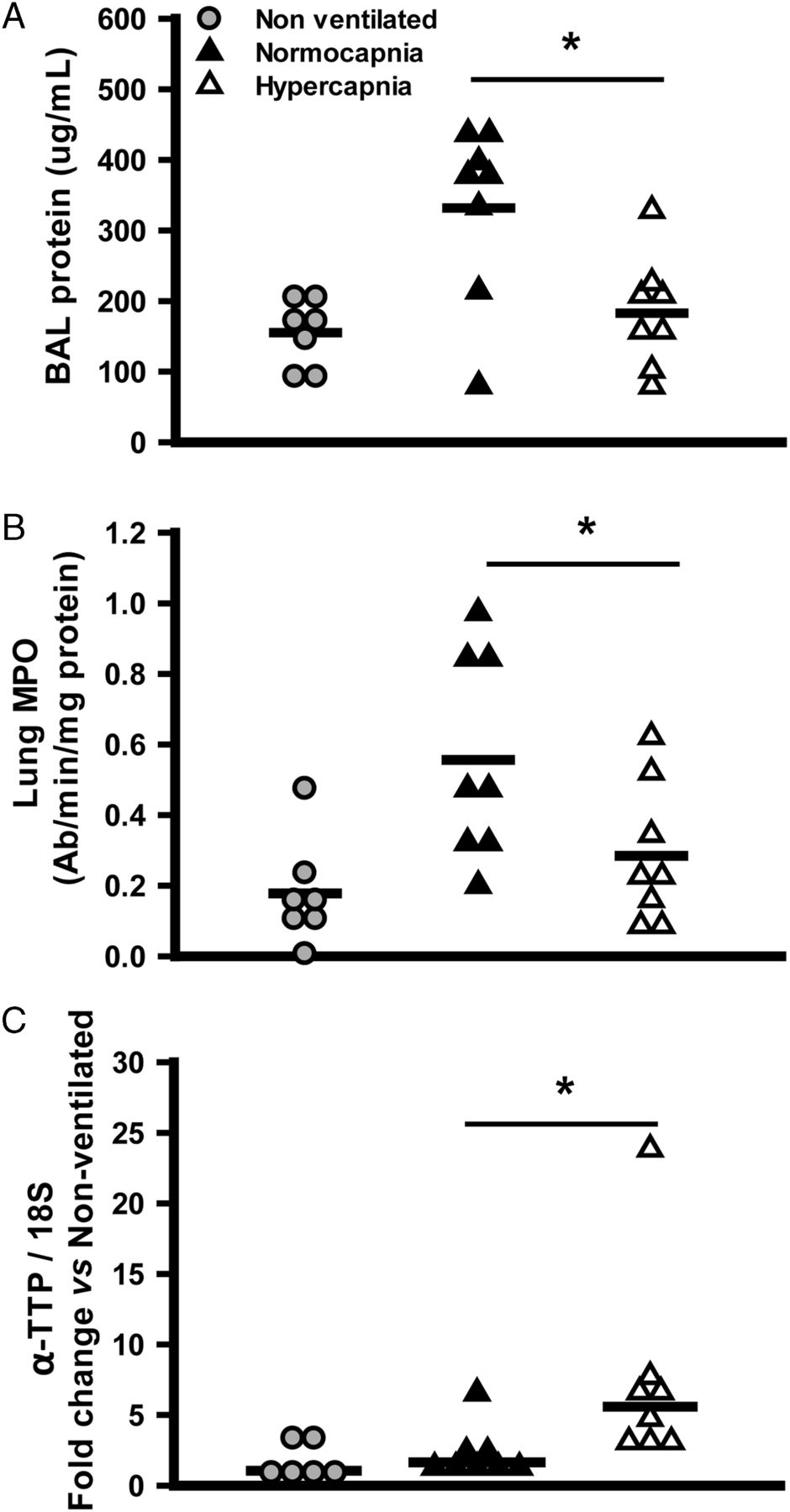
Hypercapnic protection and expression of α-tocopherol transferase protein (αTTP) in milder injury. Hypercapnia reduced protein concentration in the bronchoalveolar lavage (BAL) fluid (A), lung tissue myeloperoxidase (MPO) activity (B), and increased the tissue expression of αTTP mRNA (C), in a model of milder ventilator-induced lung injury (4 hours, VT=20 mL/kg) (*p<0.05 vs normocapnia, t test). Data from small numbers of non-ventilated mice are presented for illustrative purposes but excluded from statistical analysis.
supplementary data
The top canonical pathways associated with gene expression from high VT (figure 4A) included inflammation-related signalling pathways known to be associated with VILI (NFκB, IL-6, p38 MAPK, HMGB1). A single pathway, the NRF2-mediated oxidative stress response, was shared by high VT and by hypercapnia (figure 4B); this is a protective response against oxidative and xenobiotic toxicity, and potentially against VILI.16 High VT increased expression of NRF2 target genes, but these were decreased (not further increased) with hypercapnia (figure 5).
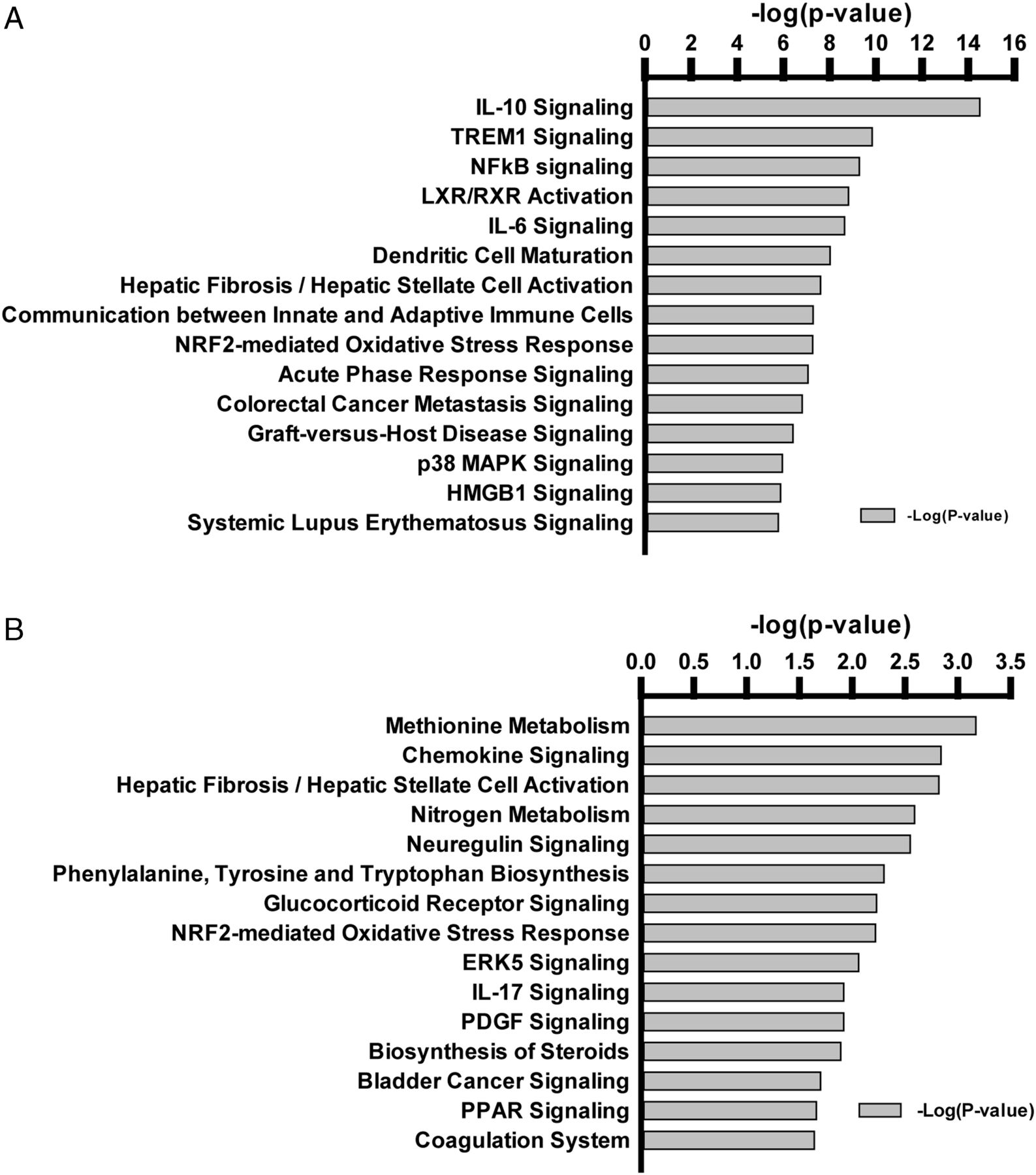
Top canonical pathways in response to mechanical ventilation. Differences in mRNAs in lungs of non-ventilated and ventilated mice were identified through microarray analysis and submitted to functional enrichment analysis using Ingenuity Pathway Analysis. Highly significant genes were selected by Fisher's exact test (at p<0.0001), and the statistical significance of each pathway is presented as negative log (p value). Pathways associated with genes affected by stretch (A) or associated with genes affected by hypercapnia (B) are illustrated.

Relative expression of mRNAs of the antioxidant response pathway in lungs of mechanically ventilated mice. NRF2 target genes heme oxygenase (HMOX1; A) and glutathione-S-transferase (GSTA1; B) were more highly expressed in high stretch normocapnia than all other ventilated groups ; that is, addition of hypercapnia reduced the expression of each during high stretch ventilation (*p<0.01 vs all other groups, one-way ANOVA).
αTTP, the most upregulated gene, is a cytosolic protein that shuttles α-tocopherol (vitamin E), an important antioxidant, between cellular membranes. We hypothesised that increased αTTP expression in hypercapnia provides antioxidant protection upstream of the antioxidant NRF2 pathway, thereby limiting its induction.
αTTP gene deletion: impact on protection from CO2
To examine the role of αTTP in hypercapnia-mediated protection from VILI, we used knockout (αTTP−/−) and wild-type sibling (αTTP+/+) mice subjected to high VT ventilation. All mice had similar lung compliance at baseline, and similar decreases in compliance over 3 hours during high VT ventilation with normocapnia (see online supplementary figure S2A). Hypercapnia attenuated several markers of injury in αTTP+/+ but not αTTP−/− mice: compliance (figure 6A, B), lung tissue MPO and TNFα mRNA (figure 7A, B), and BAL levels of MCP-1 and KC (figure 7C, D). Also, hypercapnia tended to decrease BAL IL-6 in αTTP+/+ mice only (figure 7E).

The α-tocopherol transferase protein (αTTP) gene is required for CO2-mediated protection of compliance in murine ventilator induced lung injury. High-stretch (injurious) mechanical ventilation results in decreased static compliance in αTTP+/+ and αTTP−/− mice. Hypercapnia is protective (ie, reduces the injury) in αTTP+/+ (A), but not in αTTP−/− (B) mice (*p=0.001, t test). KO, knockout; WT, wild type.

CO2-mediated protection and impact of α-tocopherol transferase protein (αTTP) on biochemical injury markers. Tissue levels of myeloperoxidase (MPO) activity (A) and TNFα mRNA (B) are significantly less after 3 hours of injurious ventilation with hypercapnia versus normocapnia (ie, hypercapnia protective) in αTTP+/+ (left panels) mice, but not in αTTP−/− (right panels) mice. Trends for these patterns (hypercapnic protection in αTTP+/+, not in αTTP−/−) were preserved for bronchoalveolar lavage levels of monocyte chemoattractant protein 1 (MCP-1; C), keratinocyte chemoattractant (KC; D), and interleukin 6 (IL-6; E) (*p<0.05 vs normocapnia, t test). Data from non-ventilated mice are presented for illustrative purposes but excluded from statistical analysis. KO, knockout; WT, wild type.
Role of αTTP and α-tocopherol in protection with hypercapnia
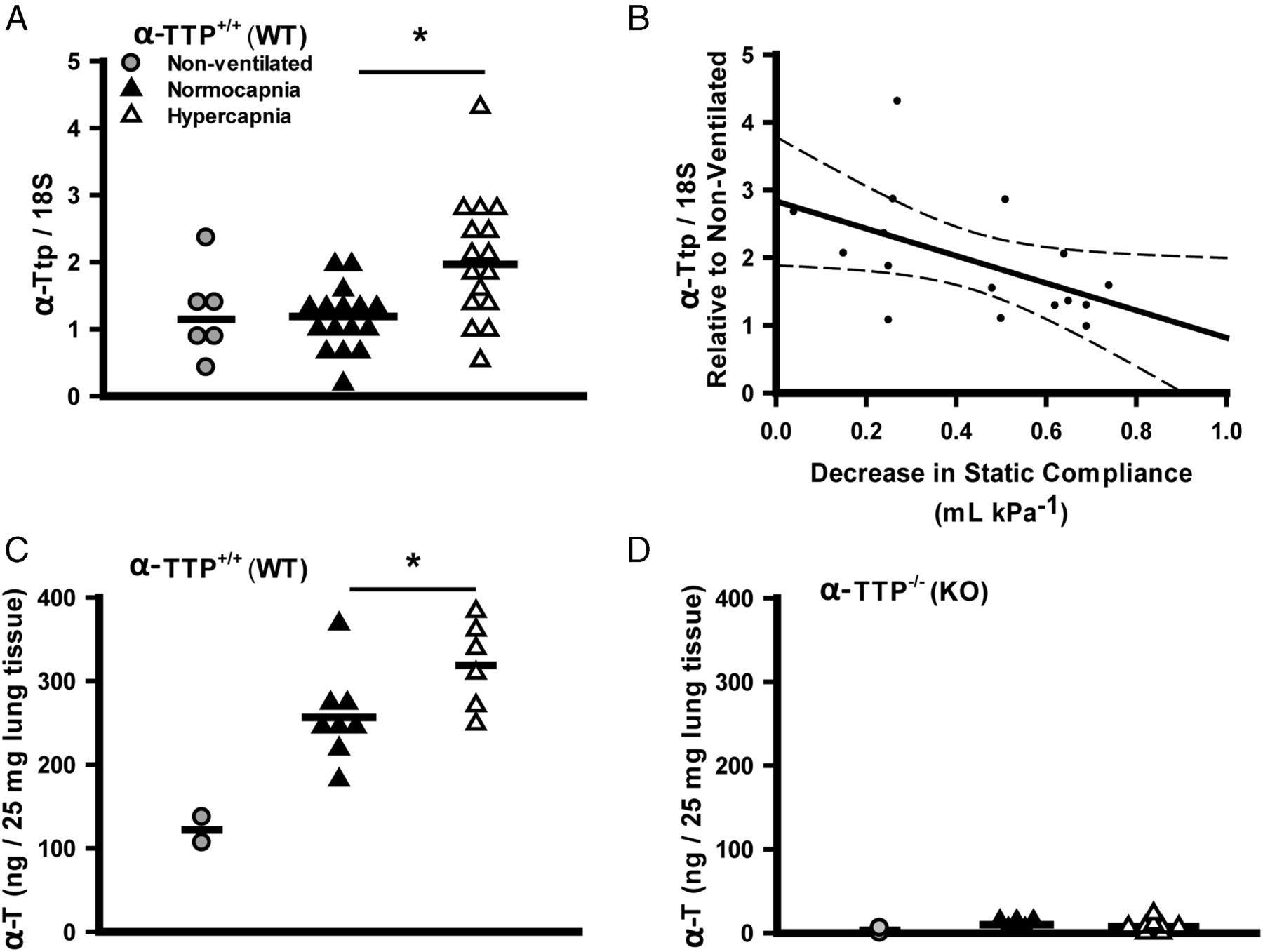
Hypercapnia induced lung tissue expression of αTTP mRNA in αTTP+/+ sibs (figure 8A) following high VT ventilation, and greater expression of αTTP mRNA was associated with greater protection (figure 8B) (as with C57BL/6J mice: table 1, figure 2). In addition, lung tissue α-tocopherol levels were increased by hypercapnia during high VT ventilation in αTTP+/+ sibs (figure 8C), while in αTTP−/− sibs, levels were 20–50 fold less and were not altered by ventilation (figure 8D).
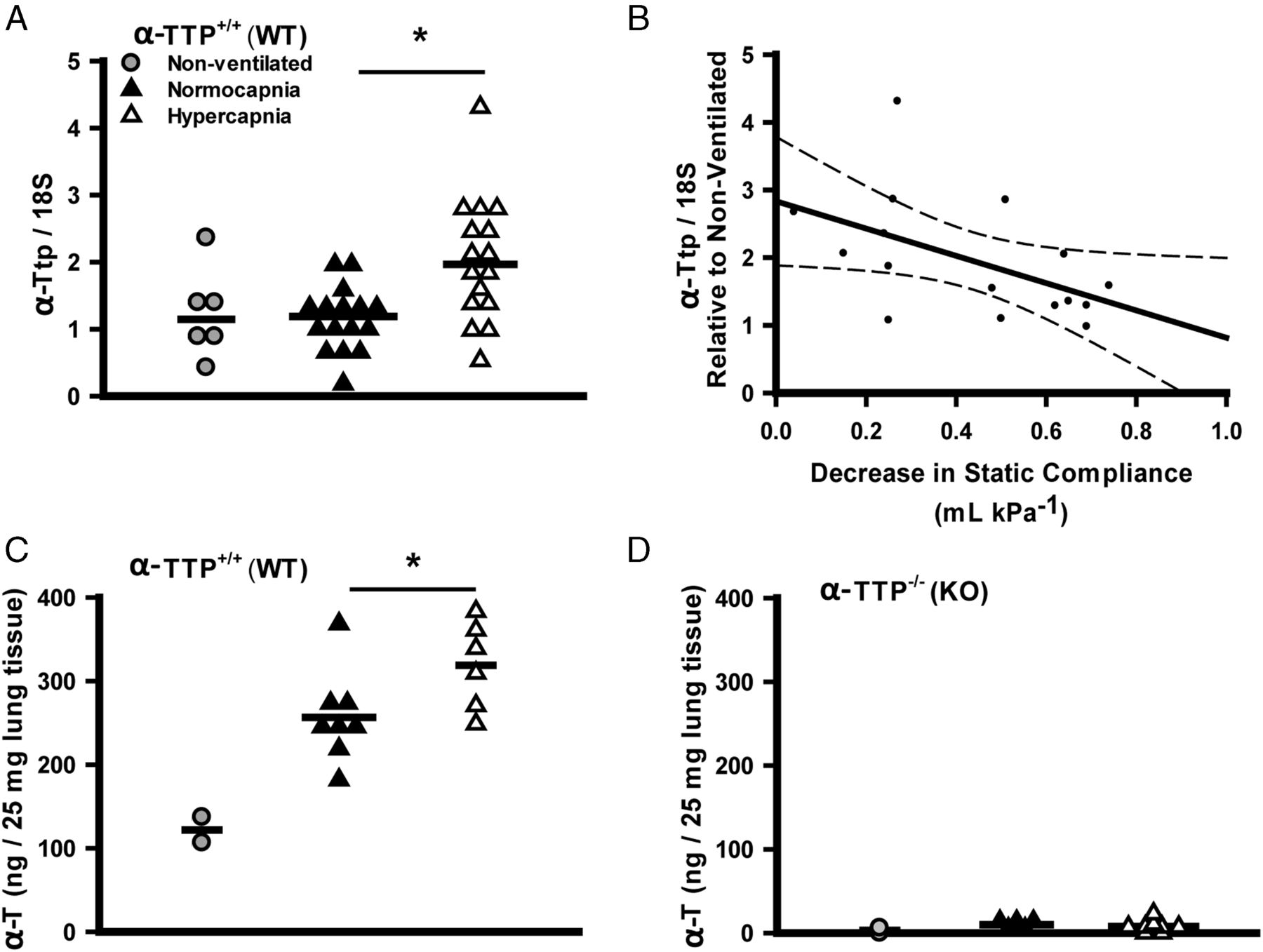
Effects of mechanical ventilation and hypercapnia on lung α-tocopherol transferase protein (αTTP) mRNA expression and α-tocopherol levels. Hypercapnia induced expression of αTTP mRNA in lung tissue of αTTP+/+ mice following high-stretch ventilation (Mann–Whitney) (A). Linear regression demonstrates the correlation between expression of αTTP mRNA and CO2 protective effect (regression expressed±95% CIs; R=−0.50, p<0.05) (B). Lung tissue α-tocopherol (α-T) levels in αTTP+/+ mice are increased by hypercapnia during ventilation (t test) (C), and are 20–50 fold less in αTTP−/− mice (D) (*p<0.05 vs normocapnia). Data from (n=2–4) non-ventilated mice are presented for illustrative purposes but excluded from statistical analysis. KO, knockout; WT, wild type.
α-Tocopherol and oxidative stress in ventilator-induced lung injury
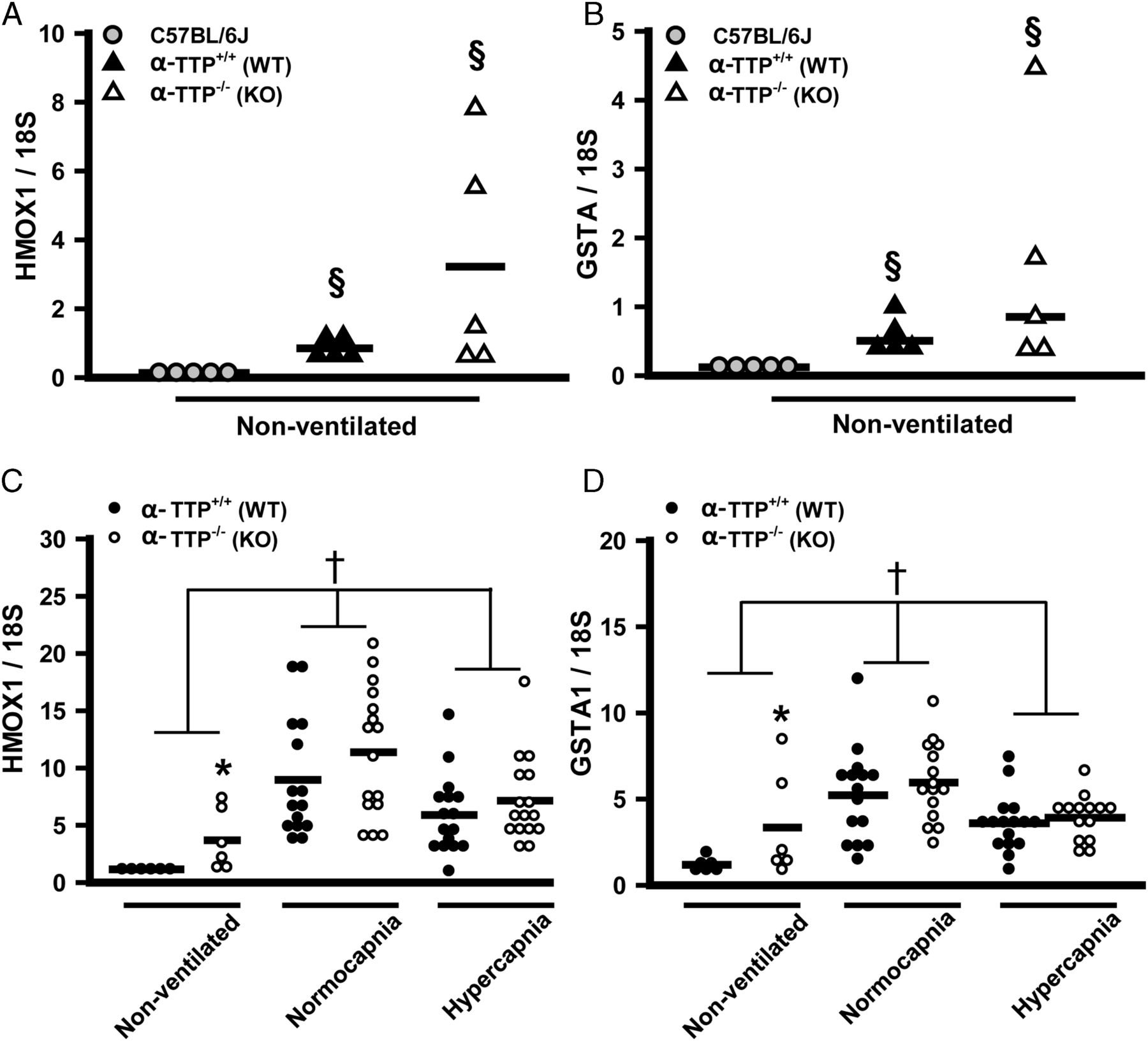
The decrease in NRF2 target gene expression due to hypercapnia in the C57BL/6J mice (figure 4B) suggested that hypercapnic induction of αTTP may exert antioxidant effects by shuttling α-tocopherol; reduced oxidative stress would then explain the lessened induction of NRF2 target gene expression. If this were true, then in the setting of lung injury, hypercapnia should lower the expression of NRF2 target genes in αTTP+/+ sibs, but not in αTTP−/− mice. Constitutive expression of HMOX1 and GSTA1 was highest in lungs of αTTP−/− (vs αTTP+/+) and lowest in C57BL/6J mice (figure 9A, B), perhaps an adaption to low α-tocopherol levels. However, αTTP−/− and αTTP+/+ mice showed similar changes in HMOX1 and GSTA1 in response to ventilation and hypercapnia (figure 9C, D), indicating that these effects are independent of αTTP expression.

Antioxidant response pathway in α-tocopherol transferase protein (αTTP+/+) and αTTP−/− murine model of ventilator-induced lung injury, effect of hypercapnia. αTTP+/+ and αTTP−/− mice expressed higher constitutive levels of mRNA for NRF2 target genes heme oxygenase (HMOX1) (A) and glutathione-S-transferase (GSTA1) (B) than C57BL/6J mice (§p<0.05 vs C57BL/6J, ANOVA on RANKS). Expression of HMOX1 (C) and GSTA1 (D) during injurious mechanical ventilation is similar in αTTP+/+and αTTP−/− mouse lungs. In non-ventilated mice, constitutive levels of HMOX1 and GSTA1 were higher in αTTP−/− versus αTTP+/+ mice (*p<0.01 vs αTTP+/+, two-way ANOVA). Both genes were similarly induced by VILI and this effect was abrogated by hypercapnia (†p<0.05 vs all other ventilation groups). KO, knockout; WT, wild type.
To determine whether αTTP protected via an antioxidant mechanism, we measured 2-hydroxyethidium as a unique marker of superoxide generation in mice injected with hydroethidine; hypercapnia had no impact on αTTP−/− or αTTP+/+ mice (figure 10A). Similar results were found using multiple markers of oxidative stress (eg, 8-isoprostane, protein carbonyls, and antioxidant capacity assay: data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hypercapnia-induced α tocopherol transfer protein (αTTP) attenuates leukotriene synthesis, not reactive oxygen species production, in murine ventilator-induced lung injury. Relative levels of superoxide in αTTP+/+ and αTTP−/− mouse lungs were measured as 2-hydroxy-ethidium (2-OH-Et, arbitrary units) in mice injected with hydroethidine and subjected to 3 hours of injurious ventilation (A). There was no significant difference in superoxide levels between normocapnia and hypercapnia in αTTP−/− or αTTP+/+ animals (Mann–Whitney). Hypercapnia suppressed levels of leukotriene B4 (LTB4, t test) and cysteinyl leukotrienes (CysLTs, Mann–Whitney) in the bronchoalveolar fluid from αTTP+/+ mice subjected to injurious ventilation, but had no effect on leukotriene levels in αTTP−/− mice (B and C). Hypercapnia did not alter arachidonic acid levels in bronchoalveolar fluid from mice of either genotype (D) (*p<0.05 vs normocapnia). KO, knockout; WT, wild type.
α-Tocopherol and eicosanoids in lung injury
α-Tocopherol inhibits 5-lipoxygenase (5-LOX),17 and its downstream products, the leukotriene B4 (LTB4) and cysteinyl leukotrienes (CysLTs: LTC4, LTD4 and LTE4), are pathogenic in lung injury and ARDS.18 Hypercapnia suppressed the BAL level of LTB4 and CysLTs in αTTP+/+, but not in αTTP−/−, mice (figure 10B, C). A similar trend was observed for the additional 5-LOX product 5-hydroxyeicosatetraenoic acid (5-HETE) (see online supplementary table S4). There were no differences in levels of the eicosanoid precursor arachidonic acid (figure 10D), or in other downstream products (eg, HETEs, prostaglandins or epoxyeicosatrienoic acids) (see online supplementary table S4).
Discussion
The current study provides evidence of a previously unsuspected mechanism by which CO2 can protect against lung injury: induction of a transporter protein for α-tocopherol, αTTP. Addition of CO2 to the inspired gas increased pulmonary expression of αTTP mRNA in different in vivo murine models of VILI; the level αTTP expression correlated with degree of lung protection; and, absence of the αTTP gene abolished the protective effects of CO2. These data provide new insights into the mechanisms of action of CO2 on molecular pathways in lung injury beyond previously described multifactorial inhibitory effects such as NFκB,8 ADAM176 and Na,K-ATPase.7 In contrast to previously described mechanisms, the current data demonstrate that CO2 upregulates expression of αTTP, which increases lung α-tocopherol levels; α-tocopherol can inhibit synthesis of the inflammatory mediator leukotrienes, products of 5-LOX which are pathogenic in acute lung injury.18
Although αTTP was required for hypercapnia's protective effects, we did not find that αTTP knockout (KO) mice injured more readily than wild-type mice in normocapnic conditions; in fact, the opposite may be true, since the decrease in static compliance in normocapnic KO mice appears slightly less than in wild-type mice (figure 6A,B). This may be due to higher constitutive levels of other protective mechanisms (eg, NRF2 responsive antioxidant genes; figure 9) as a compensatory mechanism in KO animals which have very low levels of α-tocopherol.
αTTP is a cytosolic protein that is most highly expressed in hepatocytes, where it traffics α-tocopherol and, with lower affinity, other forms of vitamin E, from endosomes to the plasma membrane in exchange for PIP2; this promotes α-tocopherol secretion into the bloodstream, bound to lipoprotein particles.19 Expression of αTTP has been described outside the liver (eg, brain, lung, prostate, placenta, retina and kidney), and while information about its non-hepatic roles is limited, some of the roles seem heterogeneous. In contrast to α-tocopherol efflux in hepatocytes,20 overexpression in prostate cells promotes accumulation of α-tocopherol,21 indicating tissue-specific functions. In addition, human mutations in the αTTP gene cause progressive ataxia and vitamin E deficiency (‘AVED’).
Surprisingly, we did not demonstrate a role for αTTP against oxidative stress in VILI, in terms of superoxide (figure 10A) or lipid peroxidation (ie, 8-Iso PGF2α, see online supplementary table S4). This may be because of higher constitutive NRF2 antioxidant expression in αTTP colony mice versus C57BL/6J mice (figure 9C, D). Indeed, an increase in constitutive expression of NRF-2-responsive genes in αTTP KO mice has been previously noted22 and may reflect an adaptation to lower levels of α-tocopherol. Our observation that WT sibs also display elevation may be due to copy insufficiency during development in a heterozygote mother. It is possible that induction of αTTP by hypercapnia in other strains (eg, C57Bl/6J) or species reduces oxidative stress.
There is nevertheless considerable evidence that vitamin E (including α, β, γ and δ tocopherols and tocotrienols) modulates the innate immune response in lung. α-Tocopherol suppresses inflammatory responses and lipopolysaccharide (LPS)-induced oxidative injury in lung epithelial cells,23 and multiple formulations of vitamin E protect in lung injury models.24 For example, in rodent models of LPS-induced lung injury, intraperitoneal α-tocopherol decreased lung oedema and neutrophil transmigration without affecting cytokine levels or NFκB activation,25 while enteral γ-tocopherol (a more powerful antioxidant than α-tocopherol) decreased neutrophils and the key inflammatory mediators MIP-2, CINC-1 and PGE2.26 A small human study demonstrated that oral γ-tocopherol reduced sputum neutrophil numbers following intranasal LPS.27
However, important gaps exist in our understanding of the protective effects of α-tocopherol: supplementation protects against lung injury in murine bacterial pneumonia (Streptococcus pneumoniae), reducing mortality, bacterial load, and neutrophil transepithelial migration.28 In addition, while α-tocopherol protects against hyperoxic lung injury through antioxidant mechanisms,29 the mechanisms of protection in other lung injury (eg, sepsis) is unclear.
Most clinical studies of α-tocopherol supplementation show minimal or no benefit in patients who have adequate dietary intake,24 including the critically ill.30 An important limitation not addressed in other studies is intracellular accumulation and trafficking of α-tocopherol in non-hepatic tissue. Increased αTTP expression could overcome such a limitation, although achieving increased expression would be a challenge. Alternatively, bypassing limited endogenous transport mechanisms by direct application of tocopherols, such as aerosolisation or incorporation into surfactant, might enhance their effects. In addition, recent data indicate that metabolites, for example, γ-tocopherol or tocotrienols, may prove more efficacious.24
Beyond neutralisation of oxygen-derived or nitrogen-derived radicals, vitamin E can regulate eicosanoid metabolism.17 Eicosanoids, including prostaglandins, thromboxanes, leukotrienes and HETEs, are an important network of signalling molecules that regulate inflammatory responses in critical illness. For example, cyclic stretch of lung epithelial cells (mimicking dyspnoea or mechanical ventilation) activates cPLA2,31 thereby providing free arachidonic acid for eicosanoid biosynthesis. α-Tocopherol modulates eicosanoid synthesis through multiple means, such as preventing the oxidation of lipid precursors, modulating mediators derived from 12-LOX and 12/15-LOX,32 and specific inhibition of 5-LOX.17 Among a panel of arachidonate products, we identified significant reduction in LTB4 and CysLTs (and a trend to decreased 5-HETE) resulting from hypercapnic induction of αTTP. Hypercapnia did not influence levels of other eicosanoids, including COX-derived prostaglandins, products of other LOX enzymes (12-HETE or 15-HETE) or non-enzymatic oxidation products of arachidonic acid (9-HETE, 11-HETE) (see online supplementary table S4). Together these data indicate that in our model, hypercapnia increased expression of αTTP; this increased accumulation and/or trafficking of α-tocopherol, leading in turn to specific inhibition of 5-LOX, and lower levels of leukotrienes.
Such lowering of leukotriene levels is a plausible downstream mechanism for protection against lung injury in the current experiments. LTB4 is a potent PMN chemoattractant, providing a key signal for transmigration into the airway mediated by specific G-protein coupled receptors (BLT1, BLT2), and CysLTs increase vascular permeability and modulate vascular smooth muscle tone via CysLT1 and CysLT2 receptors. A small clinical study has demonstrated that plasma levels of leukotrienes were elevated in patients with ARDS early in their illness, and early LTB4 levels predicted mortality from ARDS.33
In other models of lung injury, antagonism of 5-LOX is protective; for example, pharmacological inhibition and gene knockout of 5-LOX protect against murine VILI,18 and in murine sepsis KO of the 5-LOX gene (or inhibition of the enzyme) provided more protection than use of leukotriene receptor antagonists (eg, Montelukast).34 Antagonism of LTB4 is also effective in lung injury caused by haemorrhagic shock35 or in carrageenan-induced pleurisy.36
Despite preclinical success in experimental lung injury, caution is required in therapeutic targeting of leukotrienes. Many patients with lung injury have concomitant sepsis, and reduced leukotriene function could impair pathogen clearance. In addition, 5-LOX inhibition could divert arachidonic acid via alternative pathways (eg, COX), thereby altering the balance of eicosanoid signalling; in later stages this could impair production of the pro-resolving 5-LOX product, lipoxin A4. Targeting leukotriene receptors is effective in lung injury models.18 ,37 However, the high affinity LTB4 receptor, known as BLT1, is also a receptor for Resolvin E1, a pro-resolving mediator which enhances resolution of lung injury. Leukotriene A4 hydrolase (LTA4H), the enzyme that converts LTA4 (the unstable direct product of 5-LOX) into LTB4, can be targeted by several antagonists (eg, SC57461A, JNJ-26993235, or ARM1)38 to reduce LTB4 synthesis, while protecting pro-resolving mediators, but optimal blockade may also require CysLT inhibition.
There are important limitations to this study. Only male mice were used and impact of gender is unknown. We have been unable to identify the specific cell types in the lung that express αTTP due to a lack of suitable antibodies for immunohistochemistry. It is unclear whether the protective effects of αTTP (≈20% increased expression) result from the modest increase across all lung tissue, large changes in a subset of cells, or from αTTP's trafficking effect to particular subcellular locations. We have not tested whether αTTP has a protective role in other models of lung injury,3 ,4 and did not explore the mechanism by which hypercapnia upregulates αTTP expression. Finally, the αTTP gene promoter is not yet well characterised. Animal experiments investigating effects of dietary α-tocopherol and oxidative stress on hepatic αTTP expression in vivo provided conflicting data.39 However, immortalised hepatocytes upregulate αTTP in response to peroxide, hypoxia and agonists of nuclear receptors peroxisome proliferator-activated receptor α and retinoid X receptor;40 and the αTTP gene is a direct target for transactivation by the liver X receptor, a transcription factor that modulates cholesterol metabolism and lipid biosynthesis.39
While deliberate induction of hypercapnia remains an experimental intervention,2 so-called ‘permissive hypercapnia’ is widely accepted in mechanically ventilated patients; however, the case for net harm versus benefit in permissive hypercapnia is uncertain. Nevertheless, hypercapnia is an important tool in laboratory studies such as this to elucidate relevant, previously unsuspected pathways, such as upregulation of αTTP as shown here, or discovering the role of ADAM-17.6 Although the role of αTTP in lung tissue has received little attention, our data showing induction in multiple mouse models suggest broad applicability, and the correlation with lung protection is evidence of an induced, protective pathway. Targeted interventions such as aerosolisation of vitamin E formulations into the lung, or specific inhibition of LTA4H, are related to new approaches to protecting the injured lung.
Acknowledgments
This study was supported by Canadian Institutes of Health Research Operating Grant to B.P.K. (FRN 69006). BAL mediator measurements were performed by the Analytical Facility for Bioactive Molecules of The Centre for the Study of Complex Childhood Diseases, The Hospital for Sick Children, Toronto, Canada.
References
Footnotes
Contributors GO conceived the study, conducted the study, and analysed the data. DE, HA and HH participated in study design, conducted experiments, analysed data. HB, MP and BPK participated in study design, data analysis and interpretation. All authors participated in drafting or critically revising the manuscript and approved the final version.
Funding Institute of Circulatory and Respiratory Health (FRN 69006).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Primary microarray datasets are accessible through NCBI GEO (series accession number GSE86229).
Linked Articles
- Airwaves election special