Article Text

Abstract
Background Alveolar macrophages (AMFs) are critical regulators of lung function, and may participate in graft rejection following lung transplantation. Recent studies in experimental animals suggest that most AMFs are self-maintaining cells of embryonic origin, but knowledge about the ontogeny and life span of human AMFs is scarce.
Methods To follow the origin and longevity of AMFs in patients with lung transplantation for more than 100 weeks, we obtained transbronchial biopsies from 10 gender-mismatched patients with lung transplantation. These were subjected to combined in situ hybridisation for X/Y chromosomes and immunofluorescence staining for macrophage markers. Moreover, development of AMFs in humanised mice reconstituted with CD34+ umbilical cord-derived cells was assessed.
Results The number of donor-derived AMFs was unchanged during the 2 year post-transplantation period. A fraction of the AMFs proliferated locally, demonstrating that at least a subset of human AMFs have the capacity to self-renew. Lungs of humanised mice were found to abundantly contain populations of human AMFs expressing markers compatible with a monocyte origin. Moreover, in patients with lung transplantation we found that recipient monocytes seeded the alveoli early after transplantation, and showed subsequent phenotypical changes consistent with differentiation into proliferating mature AMFs. This resulted in a stable mixed chimerism between donor and recipient AMFs throughout the 2-year period.
Conclusions The finding that human AMFs are maintained in the lung parenchyma for several years indicates that pulmonary macrophage transplantation can be a feasible therapeutic option for patients with diseases caused by dysfunctional AMFs. Moreover, in a lung transplantation setting, long-term persistence of donor AMFs may be important for the development of chronic graft rejection.
- Lung Transplantation
- Macrophage Biology
- Pulmonary alveolar proteinosis
Statistics from Altmetric.com
Key messages
What is the key question?
What is the ontogeny and life span of human alveolar macrophages in vivo?
What is the bottom line?
Alveolar macrophages are self-maintained for several years in human lungs.
Why read on?
Long-term persistence of donor macrophages may be important for the development of chronic graft rejection in patients with lung transplantation.
Introduction
Alveolar macrophages (AMFs) are the prototypical macrophages (MFs) of the lungs, strategically positioned in the alveolar spaces and in the airway lumen.1 AMFs are critically important to lung function, both as phagocytes and as regulators of immune responses.2 Importantly, AMFs regulate the surfactant layer coating the alveolar epithelium, in part by clearing damaged and oxidised surfactant, and ensure low surface tension and prevent alveolar collapse.1 ,3
Like MFs residing in other tissues, including the skin, liver and gut, AMFs are functionally tailored to the highly diverse local tissue requirements,4 and this has sparked a great interest in defining the ontogeny and differentiation of MFs. AMFs have classically been assigned to the mononuclear phagocyte system,5 which comprises tissue-resident MFs derived from circulating monocytes that seed the tissues and continuously differentiate into MFs locally.6 Indeed, following AMF-depletion in experimental mice, such cells are rapidly reconstituted by blood monocytes that locally differentiate and proliferate.7 However, more recent studies using genetic fate-mapping approaches have found that murine AMFs arise from embryonic precursors that are self-maintained with little contribution from haematopoietic cells.8–11 The ontogeny and life span of human AMFs, on the other hand, is largely unresolved. However, an early study of cell smears and lung lavage from lungs of patients receiving myeloablative conditioning and haematopoietic stem cell transplantation (HSCT) to treat haematological disorders, indicated that the majority of pulmonary MFs were derived from the donor bone marrow 80 days post transplantation.12
Pulmonary alveolar proteinosis is a life-threatening respiratory insufficiency resulting from the insufficient clearance of surfactant by AMFs, and direct transplantation of MFs into the lungs has recently been suggested as a possible treatment modality for this disease,13 ,14 but more knowledge about the longevity and progenitors of human AMFs is needed to determine the therapeutic potential of such treatment in humans. Moreover, determination of the lifetime of AMFs is important to establish their potential to activate allogeneic T cells at late time points after transplantation when immunosuppression is reduced. In this study, we performed a direct temporal analysis of the turnover of AMFs in human lung transplants followed for more than 100 weeks. In addition, we analysed the development of AMFs in humanised mice reconstituted with CD34+ umbilical cord-derived cells.
Materials and methods
Patients and tissue material
We obtained transbronchial biopsies as per protocol from 10 lung transplant recipients with organs from gender-mismatched donors. The recipients comprised seven male patients, and ranged in age from 41 years to 60 years. Type of transplantation, donor and recipient characteristics as well as histological grading is summarised in table 1.
Clinical characteristics of the included individuals
The patients included were specifically selected as gender-mismatched patients transplanted from 2010 to 2011 with no evidence of rejection, according to the International Society for Heart and Lung Transplantation criteria on grading of rejection. Gender-mismatched patients in the same period with evidence of one or more acute cellular rejection requiring treatment, or patients with respiratory infection were excluded. All patients received induction with Thymoglobulin (ATG) in a dose of 1.5 mg/kg once daily for 3 days, and standard immunosuppression consisted of ciclosporin, azathioprine and low dose prednisolone as described in table 1. Surveillance bronchoscopy with transbronchial biopsies and bronchoalveolar lavage (BAL) fluid samples were performed at 2 weeks, 4 weeks, 6 weeks, 13 weeks, 26 weeks, 52 weeks, 78 weeks and 104 weeks after transplantation for detection of rejection or infection. This was performed in general anaesthesia with patients on ventilator and tubed with a laryngeal mask. All biopsies contained representative amounts of alveolar tissue, as assessed by histological examination (see online supplementary figure S1A). Additionally five open biopsies from patients with non-small cell lung cancer (three male, 50–71 years) undergoing thoracic surgery were collected. The tissue specimens were obtained distally from the tumour-affected area. All biopsies were fixed in formalin and embedded in paraffin according to standard protocols.
Supplementary figure
(A) Hemoatxylin-stained tissue section of representative area of lung parenchyma assessed in the study. (B) Compiled percentages of AMFs with identifiable chromosomes, separated on gender of the recipient.
Analysis of chimerism
Analysis was performed as described.15 Briefly, tissue sections (4 μm) were cut, mounted on Superfrost slides (Thermo Scientific) and dried overnight at 56°C. Sections were then dewaxed in xylene, and rehydrated in 70% ethanol, followed by 20 min cooking in 20 mM citrate buffer (pH 6) to denature the DNA. The specimens were subsequently incubated overnight at room temperature with 250 ng chromosome enumeration probe X/Y DNA probes (Abbott Molecular), followed by stringency washing in saline-sodium citrate/formamide according to the manufacturer's instructions. The sections were then sequentially incubated with primary and secondary antibodies for 1 hour at 37°C, counterstained for 10 min at RT with Hoechst DNA stain to visualise nuclei and mounted with polyvinyl alcohol. The antibodies for immunofluorescence staining are listed in online supplementary tables S1 and S2. Irrelevant isotype-matched and concentration-matched antibodies were used for control staining. Images were acquired with a Fluoview FV1000 confocal microscope (Olympus). To quantify the cellular chimerism, a minimum of 50 AMFs within at least 6 microscopic fields (0.045 mm2) with identifiable chromosomal configuration per section of lung parenchyma, were counted and scored as either of donor or recipient origin. The median number of cells per field was multiplied by 2222 to estimate cells/cm2. A summary of cell counts is given in online supplementary table S3.
Supplementary tables
Humanised mice
Umbilical cord blood was collected and mononuclear cells were isolated using Lymphoprep (Axis-Shield PoC) and subsequently CD34-enriched by means of MACS CD34 Microbead Kit (Miltenyi Biotec) according to the manufacturer's instructions. NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ mice (NSG) mice were acquired from The Jackson Laboratory. NSG mice 8–12 weeks of age were sublethally irradiated (3.75 Gy) up to 24 hours before intravenous injection of 105 human CD34+ cells. Lungs were harvested 10 weeks after reconstitution, fixed in formalin and processed as described above. Lungs from severe combined immunodeficiency (SCID) mice 8–12 weeks of age were obtained from Taconic, and fixed in a similar way to be used as controls. For immunofluorescence staining, sections were heat-treated in 10 mM Citrate pH 6 buffer, and stained with the antibodies listed in online supplementary tables S1 and S2.
Data analysis
All assessments were based on repeated analysis of 10 patients receiving lung transplants, and data in bar graphs represent median values of all measurements. GraphPad Prism V.5.0 (GraphPad Software, La Jolla, California, USA) was used for data processing.
Results
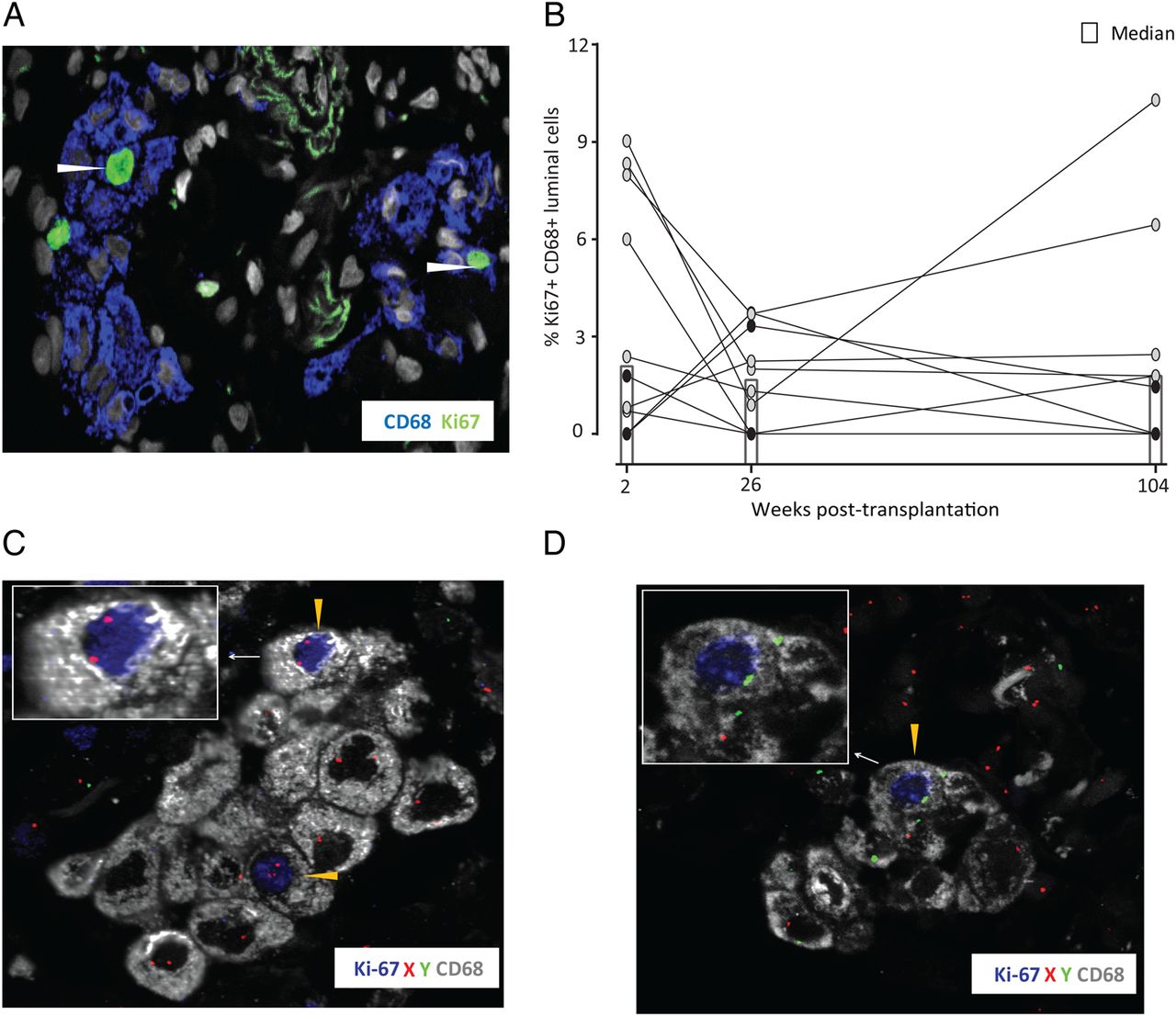
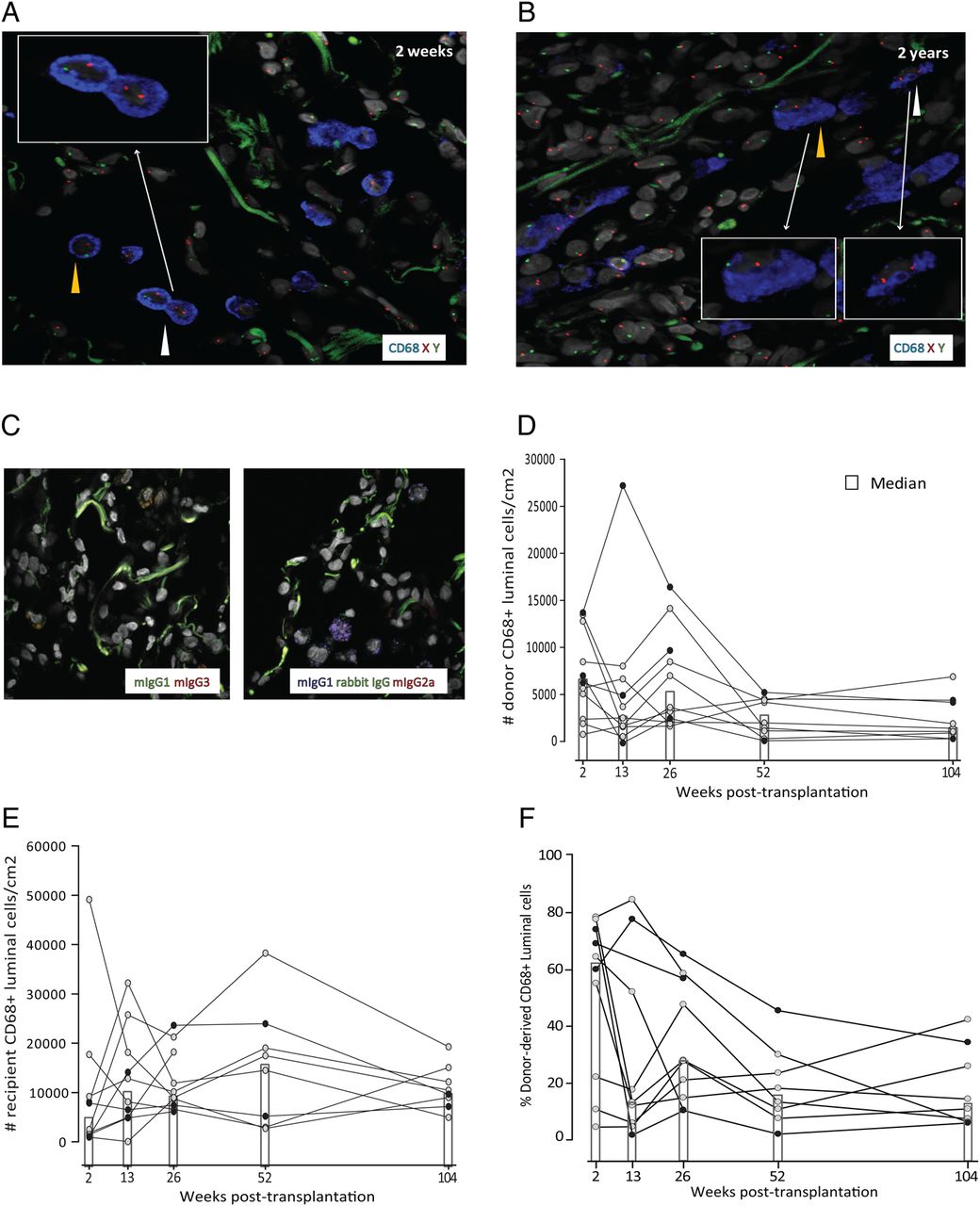
To directly determine the longevity of human AMFs in vivo, we analysed the abundance of donor-derived AMFs in transplanted lungs of patients receiving grafts from gender-mismatched donors. Transbronchial biopsies obtained at 2 weeks, 13 weeks, 26 weeks, 52 weeks and 104 weeks post transplantation were analysed by fluorescent in situ hybridisation (FISH) with X/Y probes followed by immunofluorescence staining for the well established monocyte-marker and MF-marker macrosialin/CD68. Examination by confocal microscopy provided clear identification of AMFs as large CD68+ cells confined to the alveolar lumen surrounded by the autofluorescent connective tissue (figure 1A, B). In about half of the CD68+ cells, nuclear staining of both sex chromosomes could be detected (figure 1A,B, see online supplementary figure S1B and table S3), thus enabling discrimination of donor-derived or recipient-derived AMFs. Figure 1D shows that donor AMFs were maintained in all patients during the 2-year follow-up period. Between 2 weeks and 26 weeks post transplantation the interindividual variation of cell densities was larger than at week 52 and week 104, but, importantly, there was no striking reduction in the abundance of donor-derived AMFs during the 2-year period.

Detection of donor-derived and recipient-derived alveolar macrophages in transplanted lungs. Combined X/Y-fluorescent in situ hybridisation (FISH) and immunofluorescence staining for CD68 on transbronchial biopsies taken 2 weeks (A) and 2 years (B) after transplantation from a female patient receiving a male donor graft. Yellow and white arrowheads indicate donor-derived and recipient-derived alveolar macrophages, respectively. Images are representative of 10 patients analysed. (C) Immunofluorescence staining of transbronchial biopsies using irrelevant isotype-matched and concentration-matched primary antibodies as indicated, and Hoechst DNA stain (original magnification 600×). Compiled abundance estimates of donor-derived and recipient-derived alveolar macrophages over time in gender-mismatched patients with lung transplantation in (D) and (E), respectively. (F) Percentages of donor-derived alveolar macrophages at the time points analysed. The x axis indicates weeks after transplantation. Filled dots in (D–F) indicate female recipients.
In parallel, we observed a rapid accumulation of recipient-derived CD68+ cells in the alveoli, and already at 2 weeks post transplantation 50–60% of alveolar CD68+ cells were from the recipient (figure 1E, F). The abundance of recipient CD68+ cells also showed considerable interindividual variation, but there was no striking difference between the number of recipient CD68+ cells at 2 weeks and the later time points. The interindividual variation observed did not correlate with the underlying disease or medication used for maintenance therapy (not shown). The rapid increase of recipient-derived CD68+ cells strongly suggested that blood monocytes were recruited to the transplanted lungs. We have recently demonstrated that monocytes under steady-state conditions constantly migrate to the human gut and differentiate into mature MFs (A Bujko et al, in preparation), and we have defined three stages of differentiation based on their expression of the antimicrobial protein calprotectin (S100A8/A9) and the MF marker DC-SIGN/CD20916 ,17). Monocytes that enter the intestinal mucosa express calprotectin, but not DC-SIGN. However, monocytes rapidly induce the expression of DC-SIGN and become calprotecin+DC-SIGN+ intermediates, which further differentiate into calprotecin-DC-SIGN+ mature MFs (A Bujko et al in preparation). By examining samples at 2 weeks post transplantation, we found many CD68+ cells that coexpressed calprotectin and variable levels of DC-SIGN intermingled with larger CD68+DC-SIGN+calprotectin-AMFs (figure 2A). However, at 2 years post transplantation, the alveoli contained only the latter population, and calprotectin+ monocytes were undetectable (figure 2A). To examine whether this change in phenotype was related to the transplantation procedure or immunosuppressive treatment, we phenotyped AMFs under steady-state conditions by examining unaffected lung tissue from patients with lung cancer (n=5). Similar to the 2-year samples, we found that virtually all AMFs at steady state were uniformly large, CD68+DC-SIGN+calprotectin- cells (data not shown). Together, these findings were compatible with a situation in which a large number of circulating monocytes seed the lungs within the 1st weeks after transplantation, and differentiate into long-lived AMFs, thus maintaining the abundance of AMFs with minor subsequent contribution from circulating monocytes.

Human monocytes develop into alveolar macrophages in vivo. (A) Immunofluorescence staining for CD68, DC-SIGN and calprotectin on transbronchial biopsies from gender-mismatched patients with lung transplantation 2 weeks (upper row) and 2 years (lower row) after the transplantation. White arrowheads indicate CD68+calprotectin+ monocytes with variable expression of DC-SIGN. Yellow arrowheads indicate CD68+DC-SIGN+calprotectin-alveolar macrophages. Images are representative of 10 patients analysed. (B) Immunofluorescence staining for human (h)CD68, hCD45, hCD11c and hDC-SIGN on lung biopsies obtained from humanised mice 10 weeks after adoptive transfer. Images are representative of six mice analysed (original magnification 600×).
To further substantiate the finding that monocytes were able to differentiate into AMFs in vivo, we generated humanised mice in which human CD34+ cord blood cells were adoptively transferred to NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice. The lung parenchyma of successfully reconstituted mice (n=6) was examined for the presence of human AMFs 10 weeks post transfer, by using species-specific antibodies that showed no reactivity in the lungs of control mice (not shown). Co-staining for human (h)CD45 and hCD68 showed robust populations of luminal hCD45+hCD68+ cells in the lungs of all mice analysed (figure 2B). Furthermore, hCD68+ luminal cells coexpressed hCD11c (figure 2B), which is shown to be expressed by mature AMFs,1 ,2 and were weakly positive for hCD14 (not shown), which is in line with previous reports showing that AMFs exhibit low expression of this marker.1 To further substantiate that the luminal hCD45+hCD68+hCD11c+ cells were derived from monocytes, we assessed their expression of hDC-SIGN. We found that the vast majority of luminal hCD68+ cells were hDC-SIGN+, thus further supporting that AMFs in humanised mice are derived from circulating monocytes (figure 2B).
A hallmark of mouse AMFs is their ability for local self-maintenance and repopulation following depletion through proliferation.11 ,19 We thus analysed the proliferative status of AMFs in the transplanted lungs by staining for the proliferation marker Ki67-antigen. We consistently detected a subset of AMFs expressing Ki67 (figure 3A) at all time points analysed. Although the fraction of proliferating AMFs was variable between patients, a stable median of 2% was found at 2 weeks, 26 weeks and 104 weeks (figure 3B), which is in the range of previously published data for human AMFs in healthy individuals.20 The variable number of Ki67+ AMFs observed did not correlate with the underlying disease or type of maintenance therapy (not shown). Importantly, both donor-derived and recipient-derived AMFs expressed Ki67, as assessed by combined CD68, Ki67 and X/Y-FISH analysis (figure 3C, D). Unfortunately, the low number of donor-derived Ki67+ AMFs did not allow accurate estimates of the fraction of proliferating donor cells. When determining the abundance of AMFs (both recipient and donor), we found this to be quite stable through the 2-year period, with a median range of 13 500–22 800 AMFs/cm2. To compare this with the steady-state situation, we determined the abundance of AMFs in unaffected lung tissue obtained from patients with lung cancer (n=5). A median of 49 100 AMFs/cm2 (IQR 30 000 AMFs/cm2) were found in this material. Taking into account that we only counted about half of the AMFs (those with two identifiable sex chromosomes) the AMF numbers in our transplantation material were similar to the steady-state situation.
{kind=link}
{kind=link}
{kind=link}
Both donor-derived and recipient-derived alveolar macrophages proliferate in situ. (A) Immunofluorescence staining for CD68 and Ki-67 of transbronchial biopsy obtained 2 weeks after transplantation. The arrow heads indicate proliferating alveolar macrophages (original magnification 600×). (B) Compiled proportions of proliferating alveolar macrophages over time in gender-mismatched patients with lung transplantation. The x axis indicates weeks after transplantation. Combined X/Y-fluorescent in situ hybridisation (FISH) and immunofluorescence staining for CD68 and Ki-67 of a transbronchial biopsy 26 weeks after transplantation from a male patient receiving a female graft. Yellow arrowheads indicate donor- and recipient-derived proliferating alveolar macrophages in (C) and (D), respectively (original magnification 600×). Filled dots in (A) and (B) indicate female recipients.
Discussion
Our findings strongly suggest that human AMFs are long-lived and self-maintained cells in vivo. This notion is based on the following observations: We found no significant reduction in the number of donor AMFs when analysing transbronchial samples at multiple time points for 2 years after transplantation. Moreover, a fraction of these cells were positive for the proliferation marker Ki67, demonstrating that AMFs have the capacity to proliferate locally. Although our data do not allow us to distinguish whether AMF maintenance is a result of longevity or local proliferation, the study clearly indicates that the human AMF compartment under steady-state conditions is largely independent of recruitment of cells from the circulation. At 2 weeks after transplantation, we found the abundance of donor-derived AMFs to be significantly lower than the total numbers of AMFs observed in normal lung tissue. However, several studies have shown that transplantation-related ischaemia and reperfusion cause lung injury with significant apoptotic cell death in the alveoli.21 ,22 Moreover, studies of lung transplantation in pigs reveal a large fraction of apoptotic caspase-3+ AMFs in the grafts 24 hours after transplantation (Helge Scott, personal communication). The immunosuppressive treatment given at the time of transplantation may also impact on the number of AMFs. Together, these factors may have considerable effect on the numbers of AMFs in the graft and to a large extent explain the lower number of donor-derived AMFs observed at 2 weeks. Interestingly, the reduced number of donor AMFs was fully compensated by a rapid increase in CD68+ recipient cells. In fact, the abundance of donor-derived and recipient-derived AMFs together was quite stable through the 2-year post-transplantation period, and similar to that found in normal lung tissue. This suggests that the abundance of AMFs is tightly regulated, and that sudden depletion of resident AMFs triggers rapid replacement by circulating monocytes. Factors responsible for increased monocyte recruitment after transplantation are not resolved. However, analysis of factors in BAL fluid obtained from donor lungs after ischaemia reperfusion revealed an early induction of inflammatory cytokines including interferon (IFN)-γ and interleukin 18,23 which have been shown to induce expression of the monocyte chemoattractant CCL2/MCP-1 in MFs.24
Our results suggested that recruited monocytes matured into AMFs and together with resident AMFs re-established a stable mixed chimerism where both resident and monocyte-derived AMFs are self-maintained by low-level proliferation. As there are no markers to distinguish embryonic and (monocyte-derived MFs in humans, we were unable to determine the origin of donor AMFs in our study. However, although studies in experimental mice have shown that AMFs are yolk sac-derived, these cells are partially replaced by monocytes within 1 year.10 Thus, when taking into account the obvious difference between humans and laboratory mice in terms of life span, living environment and likelihood of encountering respiratory infections, it is reasonable to assume that the donor AMFs in the transplanted lungs were of haematopoietic origin. Our results thus indicate that monocytes emigrating to the alveoli develop into self-maintained AMFs with a potential life span of several years.
This notion was further strengthened by showing that circulating monocytes potentially could differentiate into proliferating AMFs that fully compensated for the initial loss of donor AMFs following transplantation. This is consistent with recent studies in mice demonstrating that monocytes may compensate for the loss of Kupffer cells following Listeria monocytogenes infection and subsequently integrate into the self-maintaining liver MF pool when the infection is cleared.25 Moreover, cardiac MFs of embryonic origin appear to be gradually replaced with age by monocyte-derived MFs,26 and intestinal MFs are rapidly replaced by monocyte-derived MFs after birth.27 Conversely, in experimental mice with an acute lung injury induced by instillation of lipopolysaccharide (LPS) or influenza A virus,28 it was observed that a large proportion of resident AMFs persisted throughout the lung inflammation, whereas the accumulating monocyte-derived MFs progressively declined due to apoptosis following resolution. Thus, the level of monocyte-independent AMF maintenance is highly dependent on the tissue environment and the extent of cell depletion within the AMF niche. To this end, it is important to note that the complete and very rapid turnover (within 80 days) of AMFs reported by Thomas et al12 was observed in subjects receiving total body irradiation as conditioning therapy prior to HSCT. As directly shown in mice, such treatment will largely deplete the existing AMF pool, and allow repopulation of AMFs by circulating monocytes.2 Conversely, if mice lungs are shielded during irradiation, the subsequent turnover of AMFs is very low, and monocytes recruited during acute lung injury are not incorporated into the AMF compartment.28 Interestingly, however, the study by Thomas et al12 clearly shows that a number of patients exhibited a significant chimerism within AMFs several weeks after receiving HSCT, indicating the presence of persistent AMFs consistent with our study.
Our finding that human AMFs are maintained for years in vivo, lends support to the rationale of direct transplantation of monocytes-derived MFs into the lungs of patients with diseases caused by dysfunctional AMFs.13 Moreover, the benefits of lung transplantation are often undermined by chronic graft rejection, which limits the 5-year survival of transplanted recipients to about 55% and is the primary cause of death after the 1st year.29 Our results have bearing on our understanding of the underlying cellular mechanisms of graft rejection, as the long-lived donor AMFs may activate allogeneic recipient-derived T cells long after lung transplantation, when immunosuppression is reduced.
Acknowledgments
The authors thank Sara Halmøy Bakke, Kjersti Thorvaldsen Hagen and Aaste Aursjø for their excellent technical assistance.
References
Footnotes
Contributors IE-G performed all the analysis of human specimens; FLJ and ESB designed the study and wrote the manuscript; ED, CJHP and WWA generated the humanised mice; LIBS, AMH, HHLS, MI and CA selected and recruited the patients included in the study.
Funding This work was partly supported by the Research Council of Norway through its Centers of Excellence funding scheme, project number 179573 and grants from the South Eastern Norway Regional Health Authority, project number 1012105.
Competing interests None declared.
Ethics approval Norwegian Regional Committee for Medical Research Ethics, The National Committee on Health Research Ethics in Denmark and the Lund/Malmö Animal Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.