Article Text

Abstract
The pulmonary endothelium is a dynamic, metabolically active layer of squamous endothelial cells ideally placed to mediate key processes involved in lung homoeostasis. Many of these are disrupted in acute respiratory distress syndrome (ARDS), a syndrome with appreciable mortality and no effective pharmacotherapy. In this review, we consider the role of the pulmonary endothelium as a key modulator and orchestrator of ARDS, highlighting advances in our understanding of endothelial pathobiology and their implications for the development of endothelial-targeted therapeutics including cell-based therapies. We also discuss mechanisms to facilitate the translation of preclinical data into effective therapies including the application of biomarkers to phenotype patients with ARDS with a predominance of endothelial injury and emerging biotechnologies that could enhance delivery, discovery and testing of lung endothelial-specific therapeutics.
- ARDS
- Neutrophil Biology
- Pulmonary oedema
Statistics from Altmetric.com
Introduction
The pulmonary vasculature is a homogenous layer of squamous endothelial cells lining the entire pulmonary circulation. Having initially been thought of as an inert, static structure, the lung endothelium is increasingly recognised as a dynamic, metabolically active organ that modulates several key regulatory functions including: leucocyte diapedesis, intravascular coagulation, vasomotor tone, and solute and fluid trafficking via regulation of barrier permeability.
The pulmonary endothelium is distinct from the systemic vascular bed, in that it is exposed to the highest oxygen tension, while maintaining low-pressure blood flow. Coupled with the lung possessing the highest abundance of endothelial cells relative to the total cell population and its vast surface area, it is ideally placed to interact with bloodborne cells and vasoactive mediators to sense mechanical, chemical and cellular stimuli. While this allows the endothelium to regulate local (and possibly systemic) inflammation, disruption of lung endothelial homoeostatic mechanisms transforms it from a primarily anti-inflammatory phenotype to an activated proinflammatory phenotype that propagates lung parenchymal inflammation.1
Disruption of lung endothelial homoeostasis manifests clinically as acute respiratory distress syndrome (ARDS). ARDS is characterised by acute inflammation of the gas exchange surface of the lung. Dysregulated inflammation promotes the pulmonary accumulation of leucocytes and platelets, while activation of procoagulant pathways and disruption of alveolar capillary membrane barrier function leads to hypoxia, hypercapnoea and pulmonary oedema. Thus, ARDS presents clinically with acute onset of breathlessness and hypoxaemia in the presence of diffuse pulmonary oedema on the chest radiograph, with the majority of patients requiring mechanical ventilation. Risk factors for ARDS can be divided into two groups, depending on whether injury to the lung is direct such as pneumonia, with predominantly epithelial injury, or indirect bloodborne insults, such as severe sepsis, with a predominance of endothelial injury (table 1). Although mortality in ARDS has temporally declined, it remains between 25% and 35%2 and there is currently no licensed effective pharmacotherapy, highlighting the need for novel therapeutic strategies. Contemporary management focuses on treatment of the underlying cause and organ support while avoiding iatrogenic injury, most notably with low tidal volume and pressure mechanical ventilation and a conservative fluid management strategy.3
Indirect and direct ARDS—distinguishing features
In this article we focus on our current understanding of the role of the pulmonary endothelium in orchestrating and propagating ARDS, and further explore the endothelium as an emerging pharmacological target in ARDS. It is important to note that the pulmonary endothelium is structurally, morphologically and functionally distinct from the systemic vasculature (reviewed in refs. 4 ,5). Accordingly, this review will address data generated in relevant models of pulmonary endothelial injury, with reference to recent specific expert reviews where appropriate.
Structure and function of the pulmonary endothelium
The pulmonary endothelium forms a single layer of mesenchyme-derived and non-fenestrated endothelial cells. This serves as a semipermeable barrier separating the pulmonary circulation from the lung interstitium, regulating macromolecule, nutrient, leucocyte and fluid transfer.
The integrity of this barrier is determined by homophilic interactions between neighbouring endothelial cells via intercellular junctions (tight junctions and adherens junctions (AJs); reviewed in ref. 6). These junctions link endothelial cells and are served by cytoskeletal microtubules and actin microfilaments to facilitate both maintenance of barrier function and modulation of signal transduction in response to the tethering and contractile forces exerted on the endothelium during mechanical ventilation.7
Tight junctions are formed by the fusion of the outer layers of the plasma membranes and are comprised of occludins, claudins and junctional adhesion molecules coupled to cytoplasmic proteins and linked to the endothelial cell actin cytoskeleton by the zonula occludens family. AJs are composed of cadherins, primarily vascular endothelial cadherin (VE-cadherin), that bind intracellular catenin proteins (including p120-catenin, a VE-cadherin stabilising protein) that in turn bind to other protein partners in the actin cytoskeleton. AJs are mediated by calcium-dependent association of cadherin proteins and regulate the paracellular transport (the predominant pathway) of cells and solutes between the blood and the interstitium. Hence AJs, and specifically VE-cadherin, are key regulators of paracellular permeability, which determines leucocyte transmigration and oedema formation8 while cell membrane scaffolding proteins called caveolins regulate transendothelial trafficking (transcytosis) of macromolecules including albumin.9 ,10 Data suggest that transcellular permeability increases may precede and subsequently trigger paracellular permeability via Src-mediated phosphorylation of caveolin-1.10 Endothelial cells are tethered to the extracellular matrix (ECM) via interaction between cell surface integrins and their ECM ligands, which are organised in focal adhesion plaques.11
A negatively charged extracellular layer of proteoglycans, glycoproteins and glycosaminoglycans (GAGs) that line intimal surfaces, the endothelial glycocalyx, may act as an additional barrier to large molecules and circulating cells. Data from a murine ARDS model suggested that the glycocalyx-modulated neutrophil diapedesis via heparinase-mediated glycocalyx shedding and consequent exposure of neutrophil adhesion molecules,12 while in vitro human data proposed that the sialic acid component of the glycocalyx maintained barrier function via regulation of cell-matrix and cell-cell interactions.13 Despite these and other observations (reviewed in ref. 14), it remains unclear whether and how the glycocalyx contributes to the pathogenesis of human ARDS.
The endothelium performs additional regulatory roles in gas exchange, vascular tone and coagulation (reviewed in ref. 15). As an integral component of the alveolar-capillary unit, it is structurally and functionally optimised to facilitate perfusion-ventilation matching. Hence, lung endothelial cells regulate the synthesis and metabolism of vasoactive compounds such as nitric oxide and endothelin-1, potent regulators of pulmonary vascular tone. Furthermore, the endothelium also produces both prothrombic and antithrombotic substances which act both locally and remotely to regulate coagulation. It separates bloodborne cellular (eg, platelets) and humoral (eg, coagulation factors) components of the coagulation cascade from prothrombotic substances in the lung interstitium and alveolar space.
Pulmonary endothelial activation in ARDS
Pathobiology
In health, the lung endothelium adopts a predominantly inhibitory effect on inflammation and coagulation. However, upon ‘activation’ by a range of stimuli including hypoxia, cytokines (eg, tumour necrosis factor α (TNF) and interleukin (IL) 1β), chemokines (eg, IL-8), thrombin and bacterial endotoxins, including lipopolysaccharide (LPS) and interactions with activated inflammatory cells, a shift towards a proinflammatory phenotype occurs.1 Indeed, dysregulated endothelial activation and the resultant loss of homoeostatic mechanisms are aspects of ARDS pathobiology that may distinguish it from self-limiting, localised insults, for example bacterial pneumonia.1
Accordingly, lung endothelial cells are increasingly recognised as orchestrators of the inflammatory response. In experimental influenza models, the pulmonary endothelium was a key regulator of innate cellular and cytokine responses, if not the actual source of cytokine release.16 In addition, endothelial cells express various leucocyte adhesion molecules including intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and E-selectin.17 These proinflammatory responses may exhibit calcium dependency; TNF and IL-8 release from lung microvascular endothelial cells stimulated with LPS correlated with an increase in intracellular calcium,18 while cytosolic calcium oscillations induced proinflammatory gene transcription and endothelial E-selectin expression.19 Reactive oxygen species (ROS) and reactive nitrogen species production by activated cells saturate local antioxidants and contribute to tissue injury directly via downregulation of VE-cadherin,20 upregulation of neutrophil adhesion molecule expression and release of neutrophil chemotactic factors.21
Activated endothelial cells also assume a procoagulant phenotype to limit damage to lung microvasculature and localise infection. This is characterised by increased expression of platelet adhesion molecules, intra-alveolar and intravascular fibrin deposition, and release of activators of the extrinsic coagulation cascade,22 in particular nitric oxide.23 Moreover, upregulation and activation of tissue factor and loss of the ability to activate protein C and S results in capillary thrombosis and extravascular fibrin deposition, thereby contributing to the increased dead-space fraction that correlates with clinical outcomes.24
Clinical and therapeutic significance
While potentially propagating injury, the expression and release of proinflammatory molecules has driven research first into using these molecules as biomarkers of ARDS and second as putative pharmacological targets. Moreover, there is increasing evidence that the alveolar and vascular compartments are biologically distinct despite the alveolar capillary membrane disruption seen in ARDS supporting the notion that ‘phenotypical signatures’ identifying patients with site-predominant injury (endothelial vs epithelial) could be generated.
Angiopoetin-2 (Ang-2) is an endothelial growth factor, produced by endothelial cells, that regulates vascular permeability, promoting cell death and vascular regression. An incremental rise in plasma Ang-2, suggestive of progressive endothelial injury25 predicted mortality in patients with sepsis-related ARDS.26 Similarly, higher circulating GAG levels (reflecting the integrity of the glycocalyx) were found in those patients with non-pulmonary insults (ie, patients with indirect ARDS) although these did not predict outcome.27 In other studies, plasma levels of soluble thrombomodulin (TM),28 the circulating form of a transmembrane endothelial glycoprotein with antithrombotic and anti-inflammatory capabilities, and von Willebrand factor (vWF),29 a glycoprotein produced by endothelial cells, predicted mortality in ARDS. Finally, endothelial-derived microparticles (EMPs), submicron vesicles formed during membrane blebbing that shuttle proteins, organelles, lipids and RNA are an emerging biomarker of lung endothelial activation (reviewed in ref. 30), particularly in the context of mechanical stretch. Hence, EMP levels were elevated in human macrovascular endothelial cells and animals exposed to pathological mechanical stress as well as endotoxin.31 ,32
Using unbiased latent class analysis, a recent study identified two distinct cohorts of patients with ARDS who differed predominantly in their inflammatory profile and more significantly divergent responses to the application of different ventilator strategies.33 While this study did not differentiate endothelial injury from epithelial injury, it suggests that patient endotyping may hold value in predicting response to therapy. Coupled with evidence from biomarker studies outlined above, it is becoming increasingly plausible that researchers may soon be able to enrol subphenotypes of patients with ARDS with a predominance of endothelial injury to enrich enrolment in trials, thus optimising trial design and potential outcomes.
The expression of cell surface receptors and adhesion molecules also provides a putative platform to apply advances in pulmonary endothelial immunotargeting. Coupled with advanced drug delivery systems (such as liposomes, nanocarriers and host carriers) this methodology may facilitate targeting of specific aspects of lung endothelial injury in ARDS. For example, antioxidants conjugated with antibodies to the endothelial determinant Platelet/Endothelial Cell Adhesion Molecule 1 (PECAM-1) inhibited endothelial activation and reduced VCAM-1 expression in murine lung injury.34 Similarly, dexamethasone-loaded nanogels targeted to ICAM-1 accumulated in murine LPS-injured lungs and blocked expression of ICAM-1 and VCAM-1 at 24 h.35 In an effort to both target the lung endothelium and enhance biological effect, researchers have fused single-chain fragments of PECAM-1 antibodies to recombinant TM and endothelial protein C receptor (EPCR). Using this dual-targeting approach, a fivefold increase in receptor activation compared with isolated TM or EPCR targeting was observed as well as amelioration of lung injury parameters.36 Translation of these methods to clinical use will be challenging and costs may be prohibitive; nonetheless, these novel methods of targeted drug delivery hold promise.
Mechanisms of endothelial barrier disruption and injury in ARDS
Loss of barrier integrity, characterised by the formation of reversible intercellular gaps between endothelial cells, is accepted as the ultrastructural basis for the increased permeability pulmonary oedema observed in ARDS.37 A range of circulating (TNF, IL-6, LPS), released (ROS, histamine) and physical (mechanical stretch) effectors disrupt the endothelial barrier, principally by causing activation of the actin-myosin contractile apparatus which causes dispersion of cortical actin filaments and increased prominence of stress fibres which extend throughout the cytoplasm. Actinomyosin contraction of these stress fibres increases tension and is proposed to cause cell contraction, pulling cells apart and compromising barrier integrity.7 Contractile machinery is regulated by the phosphorylation status of the critical actin binding protein, myosin light chain (MLC) on Ser-19 or Ser-19/Thr-18. This is controlled through an interplay of the calcium/calmodulin-dependent MLC kinase (MLCK, phosphorylation) and Rho-regulated MLC phosphatase (MLCP, dephosphorylation).38 Hence, MLCK in particular, plays an essential role in both barrier disruption and restoration in an agonist-specific manner39–41 (figure 1).
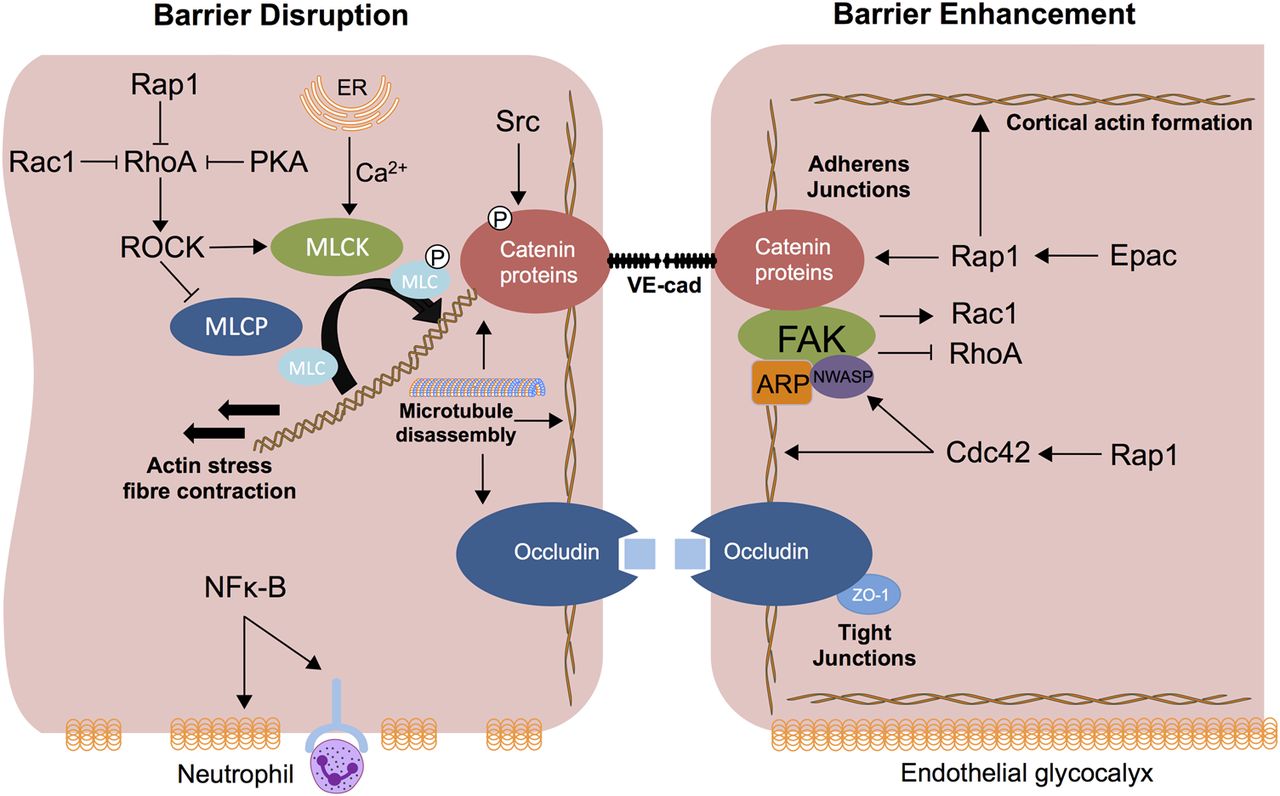
Mechanisms of pulmonary endothelial barrier disruption and enhancement. Barrier disruption results from actin-myosin interaction after MLC-phosphorylation, which is regulated by myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP). Activation of the actin myosin contractile apparatus disperses cortical actin and increases actin stress fibre formation, resulting in cell contraction and tensional force applied to adherens junction (AJ) proteins. RhoA acts via effector protein Rho-associated protein kinase (ROCK) to activate MLCK and inhibit MLCP. RhoA activity is inhibited by the GTPases Rap1 and Rac1 as well as cyclic AMP (cAMP) induced protein kinase A (PKA) activation. MLCK activation is modulated by Ca2+which enters the cytosol from endoplasmic reticulum (ER) or extracellular space. Phosphorylation of specific tyrosine residues of cytoskeletal proteins and adhesion molecules including vascular endothelial cadherin (VE-cadherin) as well as microtubule disassembly are MLCK-independent mechanisms of barrier disruption; Src mediated VE-cadherin phosphorylation leads to VE-cadherin internalisation. Nuclear factor κB (NF-κB) activation promotes a proinflammatory state resulting in degradation of the endothelial glycocalyx, which may expose neutrophil ligands. cAMP levels increase in response to a range of mediators to induce activation of PKA (which inhibits RhoA) as well as the guanine exchange factor, exchange protein activated by cAMP (Epac). Epac (via Rap1) enhances VE-cadherin junctional integrity and actin reorganisation. Rap1 enhances barrier function via inhibition of Rho and activation of Cdc42 as well as a cooperative association with VE-cadherin. Cdc42 directly regulates cortical actin organisation and proteins including MLCK and neural-Wiskott Aldrich syndrome protein (N-WASP) that mediate cortical actin formation via interaction with focal adhesion kinase (FAK) and actin-related protein (ARP) thus strengthening AJ and tight junction (TJ) formation as well as cell adhesion to the extracellular matrix (ECM). FAK also signals via effector molecules to inhibit RhoA and activate Rac1.
Although data specific to the lung endothelium is comparatively limited, regulatory small GTPases including RhoA, Rac1, cell division control protein 42 (Cdc42) and Rap1 are central intracellular regulators of the actin cytoskeleton and thus barrier function. Broadly, Rho negatively regulates barrier function42 and Cdc42 and Rap1 signalling enhance barrier function.43 ,44 RhoA through its regulated signalling circuitry, including the serine-threonine Rho-associated protein kinase (ROCK), induces phosphorylation of MLC (via MLCK) as well as inhibition of MLCP, inducing cytoskeletal remodelling and barrier permeability.45–47 Cdc42 directly regulates cortical actin organisation as well as a host of proteins including cofilin, MLCK and neural-Wiskott Aldrich syndrome protein that affect actin organisation and cell adhesion to the ECM.44 Ras-related C3 botulinum toxin substrate 1 (Rac1) can either positively or negatively regulate barrier function in a stimulus-dependent manner,48–50 while Rap1 enhances barrier function via inhibition of Rho and activation of Cdc4251 ,52 as well as a cooperative association with VE-cadherin.53
Other intracellular mediators include cyclic AMP (cAMP), nuclear factor κB (NF-κB) and focal adhesion kinase (FAK). An increase in cAMP levels in response to a range of mediators reduces vascular leakage through activation of protein kinase A (PKA) and the guanine exchange factor, exchange protein activated by cAMP (Epac).54 ,55 PKA inhibits RhoA activation and endothelial cell (EC) contraction56 and Epac (via Rap1) enhances VE-cadherin junctional integrity and actin reorganisation.53 FAK, a non-receptor tyrosine kinase regulates turnover of focal adhesion formation by binding to focal adhesion proteins as well as enhancing AJ formation;6 in experimental ARDS models (including conditional FAK deletion), decreased FAK expression was associated with lung oedema as well as albumin and neutrophil influx.57
MLC-phosphorylation independent mechanisms of barrier disruption also exist. Endothelial cell apoptosis via mediators including TNF58 and influenza virus59 may contribute. Tyrosine phosphorylation of cytoskeletal proteins and adhesion molecules including VE-cadherin, β-catenin and p120 via tyrosine kinases including Src60 may induce disassembly of the catenin-cadherin complex61 ,62 while microtubule disassembly independent of MLCK and Rho has been reported.63 ,64
The angiopoietin-Tie2 signalling axis (the endothelial tyrosine kinase Tie2 and its circulating ligands Ang-1–4) merits specific mention as a mediator of barrier disruption as it represents one of the most extensively studied barrier-regulating mechanisms. Ang-1 is constitutively expressed in a range of cell types and mediates barrier integrity and endothelial quiescence via steady activation of the Tie2 receptor, which is abundantly expressed in endothelium. Ang-2, released from endothelial cells in response to a diverse range of mediators,65 ,66 acts as a functional antagonist of Ang-1 at the Tie2 receptor, mediating cytoskeletal rearrangement25 and junctional disruption.67 Thus, mice heterozygous for Ang-2 were protected from lung injury compared with wild type mice in sepsis models.67 Ang-2 may play additional roles in leucocyte endothelial interactions.66 In the clinic, circulating levels of Ang-2 correlated with increased pulmonary oedema and mortality in patients with ARDS26 and predicted the development of ARDS in critical illness,68 further supporting a central role for Ang-2 in the endothelial injury of ARDS.
The role of damage-associated molecular patterns (native molecules released after tissue injury) and in particular mitochondrial DNA (mtDNA) production in barrier disruption is an emerging area of investigation. Hence, circulating levels of mtDNA are elevated in critical illness.69 In this context, they are potent inducers of the inflammasome via toll-like receptor 9 (TLR9),70 activating leucocyte-mediated lung injury when injected in vivo71 and endothelial barrier disruption in vitro.72 A bacterial challenge in isolated mouse lungs induced mtDNA release which was associated with endothelial hyperpermeability; this effect was replicated with exogenous mtDNA and attenuated by blockade of TLR9.73 Further elucidation of mechanisms of mtDNA release and their interplay with ROS as well as intracellular signalling pathways are required but this represents an intriguing line of investigation, if not a potential therapeutic target. While previously thought to contribute primarily to lung epithelial injury and repair,74 pathogen-associated molecular pattern (PAMP) signalling via pattern recognition receptors (PRRs) may also contribute to lung endothelial barrier dysfunction. Accordingly, influenza virus infection upregulated PRR expression (specifically TNF receptor 1) in a range of relevant models including human lung autopsy specimens, with resultant endothelial leak and apoptosis following exposure to Staphylococcus aureus-derived PAMPs in vitro.75
Candidate therapies to enhance endothelial barrier function
The endogenous lipid growth factor sphingosine-1-phosphate (S1P) enhances barrier function through a series of signalling pathways that maintain cortical actin, focal adhesions and tight junctions (reviewed in ref. 76). S1P receptors are highly expressed on pulmonary endothelial cells. Accordingly, S1P and its analogues reduced vascular leakage in small and large animal lung injury models77 ,78 as well as dampened the cytokine storm in a murine influenza model.16 The clinical application of S1P and its analogues is currently limited by systemic toxicity, most notably immunosuppression prompting the use of FTY720 (an S1P analogue) in multiple sclerosis.79 Moreover, in a murine model, prolonged application of S1P agonists worsened vascular leakage and promoted fibrosis80 while the S1P pathway has been linked to dysregulated fibrogenesis of idiopathic pulmonary fibrosis.81 Safer analogues with promising, if not superior, preclinical data may however be on the horizon.82
Based on the preclinical and clinical data outlined above, the Tie2 axis represents an attractive therapeutic target. A stable variant of Ang-1 protected against systemic microvascular dysfunction and restored endothelial barrier function in a murine sepsis model,83 while Ang-1 therapy rescued barrier disruption in vitro from ARDS plasma high in Ang-2. Recent work has demonstrated that a specific pharmacological inhibitor of VE–protein tyrosine phosphatase catalytic activity, AKB-9778, which activates Tie2,84 blocked lung neutrophil recruitment in LPS-challenged mice and stabilised lung endothelial junctions via Rap1.85 A novel Tie-2 agonist rescued mice from severe influenza up to 3 days after infection.59 Improvements in vascular leak were attributed to Tie-2-mediated attenuation of endothelial cell apoptosis as cellular proliferation was unaffected. Notably, maintenance of barrier function did not impair leucocyte transmigration. This observation supports data from a study in systemic circulation suggesting independent regulation of these two processes.86 ,87
The renin-angiotensin system (RAS) is a complex network orchestrating blood pressure, electrolyte and fluid homoeostasis, that has been implicated in the pathogenesis of ARDS. ACE2 is an endogenous modulator of RAS highly expressed in the lung endothelium and alveolar epithelium88 which diverts potentially injurious angiotensin II (Ang II) signalling via conversion of Ang II to Ang 1–7 and inactivation of angiotensin receptors, thus negatively regulating RAS.89 Studies in knockout mice have demonstrated that loss of ACE2 worsened sepsis and acid aspiration induced lung injury.90 ACE2 is reported to be the receptor for the severe acute respiratory syndrome (SARS) coronavirus induced lung injury.91 More recently, compelling data suggest that ACE2 has a central role in the development and progression of the potentially lethal avian influenza viruses H5N192 and H797.93 ,94 Gain of function ACE1 polymorphisms are also associated with ARDS susceptibility and worse outcome.95 None of these studies offer clear mechanistic insight but ACE2 signalling in the lung is likely to be mediated by alveolar epithelial cells.88 ,96 A Phase IIa trial of a recombinant ACE2 compound in patients with both direct and indirect ARDS, Clinical Trials.gov ID: NCT01597635, has recently completed recruitment and interim results are awaited.
Pleomorphic, anti-inflammatory, immunomodulatory and antioxidant effects are exerted by 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) on endothelial cells to promote cytoskeletal rearrangement, decrease oxidative stress and modulate gene expression.97 ,98 Hence, statins attenuated vascular leak in a range of murine ALI models.97 ,99 ,100 Despite promising preclinical data, including an in vivo human inhaled LPS model,101 these findings were not translated into clinical improvements in two recent clinical trials.102 ,103 Given the extensive data supporting a biological effect on the endothelium, it is intriguing to speculate that these trials may have been enriched by the enrolment of specific endotypes of patients with indirect ARDS to optimise outcomes.
A recent phase I clinical trial of 26 patients demonstrated mortality benefit in ARDS (n=37) treated with interferon-β-1a.104 The proposed mechanisms of benefit were modulation of inflammation (possibly neutrophil endothelial interactions) and endothelial barrier function via CD73-mediated dephosphorylation of AMP. While non-randomised, the mortality benefit (24% absolute reduction) in this study suggests that targeting the lung endothelium may hold promise as a viable therapeutic strategy in ARDS. A randomised controlled trial is imminent to confirm these findings. This study was also notable by the generation of human (ex vivo) data to support animal data prior to early phase trials, a paradigm that should be increasingly adopted.
The pleiotropic effects of the tyrosine kinase inhibitor imatinib, particularly in attenuation of vascular permeability induced by a broad range of mediators (discussed in105), have stimulated study into its efficacy as a barrier-enhancing agent in ARDS. Despite imatinib's association with peripheral oedema,106 case reports have suggested clinical improvements in idiopathic vascular leak107 and bleomycin-induced lung injury.108 Supporting these clinical observations these clinical observations, imatinib attenuated thrombin and histamine-induced barrier dysfunction in vitro109 and pulmonary vascular leak in clinically relevant murine models.105 ,109 Given its multiple sites of action (table 2), further mechanistic work is required to progress imatinib as a potential therapy in ARDS.
Mediators and mechanisms of barrier disruption and enhancement
Other candidate therapies have shown promise, but again mechanistic understanding in the pulmonary vasculature is not sufficiently advanced. Atrial natriuretic peptide (ANP) protects against LPS-mediated lung microvascular leakage by blocking NF-κB activity,110 while concurrently enhancing VE-cadherin localisation to AJ.111 Recent murine data support an additional role for ANP in microtubule stabilisation, an emerging mechanism in regulation of endothelial cell permeability.112 Of note, a previous small clinical trial demonstrated physiological improvements with an infusion of ANP.113 Adrenomedullin (AM), a ubiquitously expressed peptide hormone that binds calcitonin receptor-like receptor on lung endothelial cells promoting intercellular adherence, improved endothelial barrier function in preclinical acute lung injury (ALI) and ventilator associated lung injury (VALI) models.114–116 Given the abundance of binding sites on the pulmonary endothelium, AM also holds promise as a lung endothelial imaging tool using 99mTechnetium labelling.117 Finally, hepatocyte growth factor has recently been shown to suppress LPS-induced endothelial activation and barrier disruption possibly via a guanine nucleotide exchange factor.118 ,119
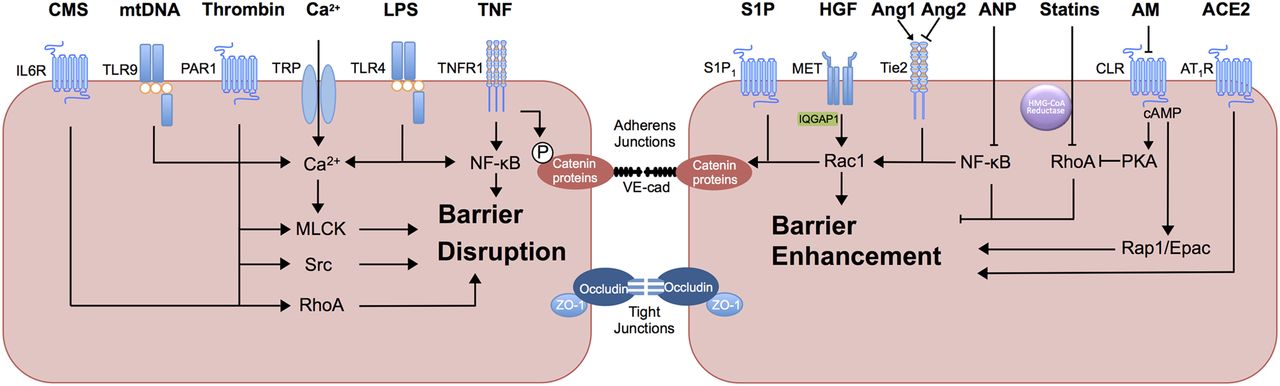
A summary of the mediators and mechanisms of barrier disruption and enhancement is outlined in table 2 and figure 2.

{kind=link}
{kind=link}
Mediators of pulmonary endothelial barrier function. Thrombin acts via protease activated receptor 1 (PAR1) to induce multiple barrier disruptive mechanisms including calcium influx, via transient receptor potential (TRP) ion channels, adherens junction (AJ) protein phosphorylation via the tyrosine kinase Src and RhoA activation. Lipopolysaccharide (LPS), via activation of toll-like receptor 4 (TLR4), increases intracellular calcium and activates myosin light chain kinase (MLCK) as well as induction of nuclear factor κB (NF-κB) signalling, promoting inflammatory cytokine production and neutrophil ligand expression. Mitochondrial DNA (mtDNA) acts via toll-like receptor 9 (TLR9) to increase intracellular calcium, activate MLCK and promote actin stress fibre formation. Cyclic mechanical stretch (CMS), via interleukin 6 receptor (IL6R) disrupts barrier function via Rho-independent mechanisms (circulating IL-6) and Rho-dependent mechanisms. Tumour necrosis factor α (TNF), via TNF receptor 1 (TNFR1) activates NF-κB. An additional mechanism of TNF-induced barrier disruption includes tyrosine phosphorylation of vascular endothelial cadherin (VE-cadherin). Barrier protective mediators sphingosine-1-phosphate (S1P) activates sphingosine-1-phosphate receptor 1 (S1P1) to promote MLC phosphorylation and AJ assembly via Rac1. S1P signalling may have additional immunomodulatory effects in influenza infection. Hepatocyte growth factor (HGF), via HGF receptor tyrosine kinase (MET) activates Rac1 activity via the adaptor protein IQGAP1. Angiopoetin-1 (Ang-1) competes with the functional antagonist Ang-2 at the tyrosine kinase with immunoglobulin-like and EGF-like domains 2 (Tie2) receptor to promote AJ assembly and cortical actin formation through Rac1 and inhibition of NF-κB signalling. Atrial natriuretic peptide (ANP) also mitigates proinflammatory NF-κB signalling and RhoA activity. 3-hydroxy-3-methylglutaryl coenzyme reductase inhibitors (statins) inhibit RhoA activity. ACE2 acts via angiotensin 2 type 1 receptor (AT1R) to inhibit the barrier-disruptive effects of Angiotensin 2 signalling and renin-angiotensin system activation. Adrenomedullin (AM) acts via calcitonin receptor-like receptor (CLR) to activate cyclic AMP (cAMP) signalling, mediating barrier enhancement via protein kinase A (PKA)-induced RhoA inhibition and endothelial cell contraction as well as Rap- mediated exchange protein activated by cAMP (Epac) activation.
Interactions of the lung microvasculature with leucocytes and platelets
Neutrophils are central to the initiation and propagation of inflammation and injury in ARDS and neutrophilic alveolitis is a histological hallmark.131 Their importance in ARDS is highlighted by the observation that a decline in respiratory function is seen in neutropenic patients with ARDS upon recovery of neutrophil counts.132 Pathway analysis of differential gene expression in sepsis-induced ARDS compared with sepsis alone identified a preponderance of genes regulating neutrophil homoeostasis and activation,133 further supporting the notion of the role of neutrophilic inflammation.
Despite extensive data from models of the systemic circulation (eg, murine cremaster vessels and human umbilical vein endothelial cells), our mechanistic understanding of the role of the pulmonary endothelium in neutrophil sequestration is limited. As a consequence of the unique pulmonary capillary microanatomy, the site and mechanisms of neutrophil sequestration are different from the systemic microcirculation. For example, owing to space constraints in the alveolar capillary bed, neutrophils must change their shape to pass through. Moreover, neutrophil rolling on the endothelial surface does not occur.134 Similarly, neutrophils exhibit selectin and CD11b/CD18-independent sequestration in pulmonary capillaries.135 ,136 A role for the endothelial glycocalyx in modulation of neutrophil adhesion molecule expression (ICAM-1) has been proposed,12 however, precise identification of the cognate receptors for neutrophils on lung endothelium remains elusive and further study in refined animal models and novel human models is an urgent unmet need.
Clarification of the role of neutrophil migration in barrier disruption is also required. Activated neutrophils exert negative effects on barrier permeability via secretion of various products such as TNF, complement component 5a and arachidonic acid but it remains unclear whether neutrophil migration through the endothelium is damaging per se. As outlined above, recent data support the concept that neutrophil migration and endothelial barrier disruption may be independently regulated.59 ,86 ,87
Recent work has focused on the role of the transient receptor potential (TRP) channels, specifically TRP vanilloid 4 (TRPV4) receptor, particularly as it is expressed on both neutrophils and the pulmonary endothelium, and specific inhibitors are available.137 TRPV4 signalling has been implicated in endothelial dysfunction secondary to mechanical138 and hydrostatic139 stress possibly via calcium influx.137 ,140 Deletion of TRPV4 attenuated both endothelial barrier leak and neutrophil activation in a murine acid injury model.137 Further, chimerical mouse models demonstrated that attenuation of lung injury was contingent on endothelial TRPV4 as opposed to leucocyte TRPV4 although deletion of TRPV on neutrophils did abrogate injury in isolated perfused mouse lungs.137
The concept that the lung endothelium may play a role in host defence by facilitating the depriming of neutrophils and hence providing protection from neutrophil-mediated remote organ injury, has been proposed. Data from an in vivo human model demonstrated that the healthy lung microvasculature retained primed cells and subsequently facilitated their depriming and release (into the systemic circulation) in an unprimed quiescent state.141 It is conceivable that failure of this mechanism, for example, as a consequence of endothelial injury in ARDS, may result in high circulating levels of primed neutrophils, which correlates with the severity of lung injury142 and which may mediate multi-organ dysfunction. Manipulation of this depriming mechanism may offer a novel therapeutic approach.
While platelets play a role in a range of pathobiological processes in ARDS (reviewed in ref. 143), our understanding of platelet endothelial interactions is underdeveloped and extrapolated from work on the systemic vasculature. In murine ARDS, platelets induced ICAM-1 expression on endothelial cells, propagating neutrophil extravasation.144 Experimental lung injury models have also demonstrated that platelets induce endothelial activation as evidenced by increased expression of vWF, P-selectin and tissue factor, such that platelet depletion and blockade of platelet binding ameliorated injury.145 ,146 Platelets also modulate endothelial barrier permeability through expression of a range of factors, including S1P (reviewed in ref. 147). Of note, however, in murine pneumonia, thrombocytopenia enhanced lung inflammation and endothelial cell activation suggesting that platelet depletion strategies may be detrimental.148 The interplay between neutrophil extracellular traps and platelets is an emerging narrative in lung injury secondary to blood transfusion149 and following lung transplantation.150 Platelet-endothelial interaction may also play a role in regulating alveologenesis (see below,151) and hence lung repair after injury.
Modulation of the coagulation cascade
Activated coagulation and depressed fibrinolysis coupled with low circulating levels of endogenous anticoagulants contribute to the pathological and physiological features of ARDS. Moreover, it is becoming increasingly apparent that inflammation and coagulation in ARDS are intimately linked.152 Modulation of these effects has thus been an attractive therapeutic target.
The protein C pathway, in particular, has been the focus of extensive research, at least in the systemic circulation (reviewed in ref. 153). Activated protein C (APC) is generated from EPCR-bound protein C by TM-bound thrombin. EPCRs are expressed on both pulmonary artery and lung microvascular endothelial cells.154 In addition to its anticoagulant activity, APC manifests a myriad of cytoprotective effects, including antiapoptotic and anti-inflammatory effects through dissociation of APC from EPCR and activation of protease-activated receptor (PAR)-1 (and to a lesser extent PAR-3) biased signalling.153 ,155 In the lung macrovascular endothelium, APC enhanced barrier function in a context-specific fashion via S1P and RAC1-dependent mechanisms.130
Despite extensive preclinical data, recombinant APC demonstrated no clinical efficacy in patients with ARDS156 and no reduction in barrier leak was demonstrated with recombinant APC therapy,157 despite attenuation of the coagulopathy.158 A multicentre trial of modified (catalytic site irreversibly blocked) recombinant factor VIIa,159 similarly, showed no benefit. Thus, we have yet to fully harness the abilities of anticoagulants to modulate cell signalling, inflammation and barrier function, in particular via PAR signalling.130 ,153 These negative trial data may temper enthusiasm for ongoing research in this area. Nonetheless, PAR-1 antagonism has recently shown beneficial effects on neutrophil migration, cytokine release and barrier disruption in a murine pneumonia model160 suggesting that alternative targets in the pathway may hold therapeutic promise.
The lung endothelium and repair in ARDS
The observation that over half of patients with ARDS survive,161 suggests that the lung microvascular endothelium and epithelium have a significant capacity for repair and regeneration. The process of repair following ARDS involves both alveolar and endothelial cell reconstitution with restoration of barrier function facilitating removal of alveolar oedema and inflammatory debris. A comprehensive discussion of endothelial repair is beyond the scope of this article and readers are referred to recent expert reviews.162 ,163
In addition to circulating progenitor cells, local populations of endothelial progenitor cells (EPCs) have been identified in the pulmonary microvascular endothelium164 and levels of EPCs are elevated in patients with ARDS.165 ,166 Retrospective analysis of lung tissue from patients with male-to-female haematopoietic stem cell transplant provides direct evidence of integration of male EPCs into female recipient pulmonary endothelium confirming the role of EPCs.167 However, the field of circulating EPCs remains controversial; their origin and function as well as their ability to effect repair under the hostile environment of clinical ARDS remains uncertain. Moreover, the cell most commonly studied, (identified by Asahara in 1997168) is now recognised as a monocyte with angiogenic features and not a true endothelial progenitor.169 Furthermore, it has been demonstrated that the majority of endothelial repairs, at least after endotoxin-induced lung injury, was affected by tissue-resident progenitor cells, not circulating EPCs.170 Any therapeutic role of cell-based therapies may be anti-inflammatory, via secretion of paracrine factors, as opposed to mediating endothelial repair per se.
Alternatively, endothelial cells may modulate neoalveolarisation via crosstalk with their local niche. Hence, in murine pneumonectomy models platelet-endothelial interaction via stromal-cell derived factor-1151 and lung endothelial vascular endothelial growth factor signalling (via the production of matrix metalloproteinase 14)171 induced alveologenesis. Similarly, lung endothelial cells were required to support alveolar stem cell differentiation in vitro and lung regeneration in vivo; this mechanism was thrombospondin-1 (an endogenous inhibitor of angiogenesis) dependent.172
These data provide compelling evidence that the pulmonary endothelium participates in the resolution of ARDS. Whether manipulation of these endothelial niches or the administration and mobilisation of EPC populations is a feasible goal in ARDS requires further study. Further definition of the molecular pathways that regulate the crosstalk between endothelial cells and the reparative niche, particularly in the context of relevant ARDS models will be invaluable. While primarily directed towards resolution of alveolar epithelial inflammation and injury, data from a clinical trial of mesenchymal stem cells173 (ClinicalTrials.gov: NCT02097641) will hopefully provide additional insights, particularly regarding the effects of anti-inflammatory strategies on endothelial barrier function and a potential interplay with endothelial cells.
Future directions
The identification and validation of robust biomarkers to phenotype patients with ARDS to either identify them as having a predominance of endothelial injury or to predict response to treatment would appear an integral component influencing the success of future trials of novel endothelial therapeutics in ARDS. The application of ‘omics’ technologies such as metabolomics (reviewed in ref. 174) will hopefully advance this field in the coming years facilitating personalised therapies within the next decade. Biomarker exploration will ideally also result in the identification of novel druggable targets. Building on the work by Calfee et al,27 ,28 ,33 ,175 enrichment of patient enrolment into focused clinical trials of novel endothelial-specific therapeutics using established biomarkers would appear to be a reasonable strategy to optimise patient outcomes.
Further, it is imperative that the heterogeneity between systemic and pulmonary vascular endothelium and between animals and humans is increasingly recognised. Developing better techniques to interrogate neutrophil-endothelial interactions in the lung would seem paramount; the continued development of live imaging in transgenic mice using fluorescent probes to label specific cell types176 and increased application of isolated perfused lung models177 as well as human ex vivo and in vivo models represent mechanisms to maximise the translation of preclinical data into effective therapeutics. It remains to be seen whether the so-called ‘lungs on a chip’,178 which combine biomimetic systems containing microfluidic channels lined by living human cells, will develop into tractable models for lung biology per se or specifically ARDS pathobiology. An IL-2-induced injury model recapitulated salient features of in vivo ARDS, which were attenuated by a novel TRPV4 antagonist.179 This technology has the potential to complement (if not replace) current ARDS research platforms, facilitating interrogation of hitherto underdeveloped aspects of lung endothelial biology and pharmacology in a coculture platform with biologically relevant cell types (alveolar epithelial cells, lung microvascular endothelial cells and leucocytes or platelets) exposed to physiologically relevant mechanical forces via a stretchable porous membrane.180 The applications of three-dimensional bioprinting for the study of lung biology181 similarly represents an enticing, if yet unrealised, prospect. Development of such innovative, humanised models coupled with the refinement of existing models will be central to the identification and testing of emerging therapeutics.
Conclusions
The pulmonary endothelium is increasingly seen as pivotal in both the progression and the resolution of ARDS and is therefore primed as a therapeutic target. Our understanding of endothelial biology, notably neutrophil-endothelial interactions in the lung vasculature, is a limitation to potential progress. Despite this, enhancement of endothelial barrier function, in particular, shows promise in preclinical models of ARDS and relevant late-phase human trials are imminent. Novel imaging techniques and innovative in vitro research platforms may facilitate translation of promising animal data into efficacious endothelial-specific therapies. Despite enthusiasm for their introduction, the use of cell-based therapies requires further characterisation of their phenotype, efficacy and safety. In the interim, the application of emerging technologies will assist the search for robust biomarkers of endothelial injury, which will inevitably enhance enrolment into future clinical trials of novel lung endothelial-targeted agents.
Acknowledgments
This work was supported by the Royal Brompton and Harefield NHS Foundation Trust, NIHR Respiratory Biomedical Research Unit, London, UK.
References
Footnotes
Twitter Follow Alastair Proudfoot at @ICUDocAP
Collaborators Stefan Jovinge.
Funding CS is a Wellcome Trust Postdoctoral Clinical Research Training Fellow (WT101692MA).
Competing interests CS was a co-investigator on a project grant funded by GlaxoSmithKline, which undertook preclinical assessment of the effects of a potential ARDS therapy on human neutrophils (2012–2014). MJG and CS have received consultancy fees from GlaxoSmithKline. MJG and AGP have had unrestricted project grant support from GlaxoSmithKline.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves