Article Text

Abstract
Background Exacerbation in asthma is associated with decreased expression of specific host defence peptides (HDPs) in the lungs. We examined the effects of a synthetic derivative of HDP, innate defence regulator (IDR) peptide IDR-1002, in house dust mite (HDM)-challenged murine model of asthma, in interleukin (IL)-33-challenged mice and in human primary bronchial epithelial cells (PBECs).
Methods IDR-1002 (6 mg/kg per mouse) was administered (subcutaneously) in HDM-challenged and/or IL-33-challenged BALB/c mice. Lung function analysis was performed with increasing dose of methacholine by flexiVent small animal ventilator, cell differentials in bronchoalveolar lavage performed by modified Wright-Giemsa staining, and cytokines monitored by MesoScale Discovery assay and ELISA. PBECs stimulated with tumour necrosis factor alpha (TNF-α) and interferon gamma (IFN-γ), with or without IDR-1002, were analysed by western blots.
Results IDR-1002 blunted HDM challenge-induced airway hyper-responsiveness (AHR), and lung leucocyte accumulation including that of eosinophils and neutrophils, in HDM-challenged mice. Concomitantly, IDR-1002 suppressed HDM-induced IL-33 in the lungs. IFN-γ/TNF-α-induced IL-33 production was abrogated by IDR-1002 in PBECs. Administration of IL-33 in HDM-challenged mice, or challenge with IL-33 alone, mitigated the ability of IDR-1002 to control leucocyte accumulation in the lungs, suggesting that the suppression of IL-33 is essential for the anti-inflammatory activity of IDR-1002. In contrast, the peptide significantly reduced either HDM, IL-33 or HDM+IL-33 co-challenge-induced AHR in vivo.
Conclusion This study demonstrates that an immunomodulatory IDR peptide controls the pathophysiology of asthma in a murine model. As IL-33 is implicated in steroid-refractory severe asthma, our findings on the effects of IDR-1002 may contribute to the development of novel therapies for steroid-refractory severe asthma.
- asthma
- airway epithelium
Statistics from Altmetric.com
Key messages
What is the key question?
Can immunomodulatory innate defence regulator (IDR) peptides alleviate airway inflammation and hyper-responsiveness?
What is the bottom line?
Synthetic immunomodulatory peptide IDR-1002 reduces allergen-induced airway inflammation and hyper-responsiveness, and suppresses the production of the cytokine interleukin-33.
Why read on?
As IDR peptides exhibit the potential to control both inflammation and infections, IDR peptides are attractive candidates for the development of new therapies for chronic inflammatory respiratory disease such as asthma, including steroid-refractory severe asthma.
Introduction
Asthma is a chronic respiratory disease characterised by airflow obstruction, bronchial hyper-responsiveness and airway inflammation.1 It is primarily driven by exposure to inhaled allergens such as house dust mite (HDM; Dermatophagoides sp).2 Despite the availability of widely used asthma therapeutics, namely glucocorticoids and β2-agonists, the disease is not controlled in ~10% of patients.3 Moreover, the available therapies including inhaled corticosteroids may contribute to an increased risk for infections.4–7 These challenges highlight the need for the development of alternate strategies that can control asthma.
Expression of specific immunomodulatory cationic peptides known as host defence peptides (HDPs) is decreased in the lungs during airway inflammation and asthma.8 9 HDPs in the lungs are primarily secreted by epithelial cells and immune cells such as neutrophils, macrophages and T-cells.10 HDPs exhibit a wide range of immunomodulatory functions which include induction of anti-inflammatory cytokines, control of endotoxin-mediated and proinflammatory cytokine-mediated inflammation and influencing the maturation and differentiation of dendritic cells and T-cells.11–15 HDPs can control both infections and inflammation13 15–18 and play a role in maintaining immune homeostasis.16 19 Glucocorticoids also significantly decrease the levels of pulmonary HDPs, which contribute to decreased ability to resist infections.9 Synthetic derivative peptides of HDPs, known as innate defence regulator (IDR) peptides, are beneficial in controlling infections and inflammation, and the therapeutic potential of these have primarily been explored in various infection models.20–22 In this study, we examined the effects of exogenous administration of an IDR peptide, IDR-1002, in a HDM-challenged murine model of allergic asthma, and further interrogated the peptide activity in human primary bronchial epithelial cells (PBECs).
Peptide IDR-1002 (VQRWLIVWRIRK-NH2) is a 12-amino acid cationic peptide derived from a bovine cathelicidin HDP, Bac2A.20 IDR-1002 attenuates inflammatory cytokine production in cystic fibrosis airway cells, exhibits antibiofilm activity and controls multidrug-resistant bacterial infections.23 24 In this study, we provide the first evidence to show that subcutaneous administration of IDR-1002 significantly reduces airway hyper-responsiveness (AHR) and leucocyte infiltration to the lungs in a HDM-challenged murine model of asthma. Our mechanistic studies show that the peptide suppresses the production of the cytokine interleukin-33 (IL-33) in murine lungs and human PBECs. We demonstrate that inhibition of IL-33 by IDR-1002 is essential to control airway inflammation, and that the peptide reduces both HDM-induced and IL-33-induced AHR in vivo. As IL-33 is a critical steroid-resistance mediator,25–28 our finding on the effects of IDR-1002 may contribute to the development of new therapies for steroid-refractory asthma.
Materials and methods
Murine models of allergic asthma and IL-33 challenge
The HDM-challenged murine model protocol used in this study is based on our previous study29. Experimental design for the animal studies and reporting of data were compliant with the ARRIVE guidelines in the reporting of in vivo animal research. Female BALB/c mice were obtained from the Genetic Modeling of Disease Center (University of Manitoba) and housed in the central animal care facility at University of Manitoba. Animals were randomly sorted into cages by the animal care facility staff, and there was a maximum of five mice per cage in all experiments. Briefly, female BALB/c mice (6 to 8 weeks old) were challenged with intranasal administrations of 35 µL (0.7 µg/mL saline) of HDM protein extract (Greer Laboratories, North Carolina, USA), five times a week for 2 weeks. IDR-1002 (CPC Scientific, California, USA) dissolved in saline was administered (subcutaneously) at a dose of 6 mg/kg per mouse 3 days per week (online supplementary figure 1A). Recombinant murine IL-33 (BioLegend, California, USA) dissolved in saline was administered intranasally where indicated, at a dose of 1 µg/mouse, in the last 5 days in the HDM-challenged mice (online supplementary figure 1B). In experiments with IL-33 challenge alone, recombinant murine IL-33 was administered intranasally, at a dose of 1 µg/mouse, for 5 consecutive days30 (online supplementary figure 1C). Mice were sedated using isoflurane prior to any intranasal instillation. HDM and IL-33 challenge, as well as peptide administrations were performed in the morning between 10:00 and noon. Mice were visually monitored for grooming and activity levels every day. Mice were sacrificed and samples were collected 24 hours after the last HDM challenge based on our previous study29 or after the last IL-33 challenge.30
Supplementary file 1
Bronchoalveolar lavage fluid (BALF) cell differential assessment
Mice were anaesthetised with sodium pentobarbital and lungs were washed with 1 mL of cold saline twice. BALF obtained was centrifuged (1200 rpm, 10 min) and cell differentials were assessed using a modified Wright-Giemsa staining (Haema 3 Stat Pack) using a Carl Zeiss Axio Lab A1 (Carl Zeiss, USA) for imaging.29 Cell differentials were counted blinded by two different personnel.
AHR measurements
Mice were anaesthetised with sodium pentobarbital, tracheostomised and lung function was measured using a flexiVent small animal ventilator (SCIREQ, Quebec, Canada) for quantitative assessment of airway constriction and stiffness of the lung, as previously described.29 31 32 Briefly, we used high-frequency forced oscillation with positive end-expiratory pressure of 3 cmH2O to assess Newtonian resistance (Rn) to monitor central airway constriction, tissue damping (G) as an index of alveolar tissue restriction and tissue elastance (H) to determine alveolar tissue stiffness. A muscle paralytic agent was not used in this method. Data were collected using flexiWare software and transferred to Excel software for further analysis. Changes in Rn, G and H were monitored in response to nebulized saline (baseline measures), followed by increasing concentrations of nebulized methacholine (3–50 mg/mL), using Quick Prime-3 and Snapshot perturbations.
Histology
Lungs were inflated in situ through a tracheal cannula, fixed with ~1 mL of 10% (v/v) formalin and dehydrated using ethanol and xylene as previously described by us.29 Paraffin-embedded lungs sectioned (6 µm) and stained with periodic acid Schiff (PAS) (Sigma) to assess goblet cell hyperplasia. A Carl Zeiss Axio Lab.A1 microscope was used for imaging along with Zen 2.3 software for analysis. Histological assessment was performed blinded by two different personnel. Circumference of airways was obtained using the Zen 2.3 (Carl Zeiss) software.
Assessment of cytokine levels
The bottom right lobe of murine lung was collected in Protein Extraction Reagent T-Per (Thermo Fisher Scientific, Rockford, Illinois, USA) containing protease inhibitor cocktail (PIC; Sigma) and homogenised on ice using the Cole-Parmer LabGEN 125 homogenizer (Cole-Parmer Canada, Quebec, Canada). Lungs were collected after AHR assessment and BALF collection. Homogenates were centrifuged at 10 000×g to obtain tissue lysates. Total protein concentration was determined using a bicinchoninic acid (BCA) assay (Thermo Fisher Scientific). Production of a panel of cytokines (interferon gamma (IFN-γ), IL-1β, IL-10, IL-12p70, IL-2, IL-4, IL-5, IL-6, keratinocyte chemoattractant (KC) and tumour necrosis factor alpha (TNF-α)) was monitored using the multiplex MesoScale Discovery assay, according to manufactures instructions. Cytokines IL-33, IL-25, thymic stromal lymphopoietin (TSLP), macrophage-derived chemokine (MDC) and thymus and activation regulated chemokine (TARC) were monitored by individual ELISA assays (R&D Systems, Minnesota, USA).
Cell culture
Human PBECs were isolated from resected lung tissue from four anonymised donors (n=4) undergoing surgery at the Leiden University Medical Center and cultured as previously described.33 Demographics of the donors are shown in online supplementary table S1. Briefly, bronchial rings were enzymatically digested with 1× Proteinase XIV (Sigma) at 37°C. Epithelial cells were isolated and cultured in keratinocyte serum-free medium (Gibco) supplemented with epidermal growth factor (EGF), bovine pituitary extract (BPE) and isoproterenol, and an antibiotic mix containing of penicillin, streptomycin and ciprofloxacin until almost confluent. Cells were trypsinized and stored in liquid nitrogen until further use. Cells were seeded at a density of 5000 cells/cm2 in tissue culture plates precoated with 30 µg/mL PureCol (Advanced BioMatrix, California, USA), 10 µg/mL fibronectin (SanBio, The Netherlands) and 10 µg/mL bovine serum albumin (BSA; Sigma) containing 1:1 mixture of Dulbecco’s modified Eagle’s medium (Gibco) and bronchial epithelial growth medium (SanBio), supplemented with 1 mg/mL BSA (Sigma), 15 ng/mL retinoic acid (Tocris, UK) and 100 µg/mL penicillin/streptomycin (Lonza). Cells were cultured until ~80% confluent, and medium replaced every 48 hours. Cells were starved in medium with no growth factors for 24 hours before stimulation. Peptides IDR-1002 or IDR1 (20 or 40 µM) were added 30 min prior to stimulation with 20 ng/mL TNF-α (PeproTech, New Jersy, USA) and/or 30 ng/mL IFN-γ (R&D Systems). Cell lysates were collected to assess protein abundance by western blots after 24 hours of stimulation.
Western blots
Human PBECs were scraped and collected in phosphate-buffered saline (PBS) containing PIC (Cell Signaling Technology, Massachusetts, USA) and centrifuged at 300×g for 7 min. The cell pellets were lysed in PBS containing PIC and 0.5% NP40 (Sigma, Missouri, USA). Samples were subjected to one freeze thaw cycle prior to centrifuging at 10 000×g for 10 min to obtain cell-free lysates. Total protein concentration was determined using a micro BCA assay (Thermo Fisher Scientific, Massachusetts, USA). Equal amounts of protein (20 µg) were resolved on 4%–20% Mini-PROTEAN TGX Precast Protein gels (Bio-Rad, California, USA), followed by transfer to nitrocellulose membranes (Millipore, Massachusetts, USA). Membranes were blocked with Tris-buffered saline (TBST) (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1% Tween-20) containing 5% milk powder. The membranes were probed with antibodies for IL-33 (Abcam, UK), IRF1 (Cell Signalling Technology), IP10 (Cell Signalling) and β-actin (Cell Signalling Technology), in TBST containing 1% milk powder. Affinity-purified horseradish peroxidase-linked secondary antibodies (Cell Signalling Technology) were used for detection. Membranes were developed using Pico and Femto ECL detection system (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Statistical analyses
Two-way repeated measures analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used to compare the means between multiple groups for lung function analyses to examine changes in Rn, G and H with increasing concentrations of methacholine. Mann-Whitney U test was used to compare PC50 values between the HDM-treated and naive groups, and between HDM-treated mice and HDM-challenged mice treated with peptide IDR-1002. In animal studies, for immune cell infiltration, cytokine levels and goblet cell counts, one-way ANOVA with Tukey’s multiple comparisons test was used for statistical analyses. Repeated measures one-way ANOVA with Fisher’s least significant difference was used for statistical analysis of western blot densitometry for IL-33, IP10 and IRF1 using human PBECs. GraphPad PRISM 6 was used for statistical analyses. Differences at a value of p<0.05 were considered to be statistically significant.
Results
IDR-1002 reduces AHR in HDM-challenged mice
Previous studies have shown that HDM challenge (intranasally) for 2 weeks in mice results in AHR and airway inflammation.29 34 Therefore, we used this preclinical model to examine the effects of exogenous administration (subcutaneously) of IDR-1002 (6 mg/kg per mouse). The route and dose of administration of IDR-1002 were based on previous studies.20 21 Assessment of lung mechanics showed that administration of IDR-1002 significantly reduced both maximum methacholine (50 mg/mL)-induced Rn by 56%±31% (figure 1A) and decreased sensitivity to methacholine (amount required to increase the baseline airway resistance by 50% (PC50)) by 2-fold±0.47-fold (figure 1D), compared with HDM-challenged mice. IDR-1002 restored methacholine PC50 in HDM-challenged mice to levels similar to that in allergen-naive mice (figure 1D). We observed concomitant inhibition of HDM challenge-induced maximum tissue damping (G) by 49%±34% and tissue elastance (H) by 50%±37% with IDR-1002 treatment (figure 1B,C respectively), at maximal methacholine concentration. Notably, at lower concentrations of methacholine (6–12 mg/mL) in HDM-challenged mice, IDR-1002 fully abrogated increased Rn, G and H to similar levels seen in allergen naive animals. Taken together, these results indicate that the peptide exhibits the ability to decrease overall resistance of the airway tree (Rn), tissue resistance in the alveoli (G) and tissue elastance (H), indicating that the peptide has significant functional impact on airflow conductance of central, small and terminal bronchioles. Administration of IDR-1002 alone had no effect of indices of airway resistance and lung function in allergen-naive mice (figure 1). These results demonstrate that administration of IDR-1002 significantly reduces AHR in allergen-challenged mice.

IDR-1002 reduces HDM challenge-induced airway hyper-responsiveness in mice. Female BALB/c (8–10 weeks) mice (n=9 per group) were challenged with 35 µL of whole HDM extract (0.7 mg/mL) in saline intranasally for 2 weeks. IDR-1002 was administered subcutaneously (6 mg/kg) three times a week. Lung mechanics was monitored using a flexiVent small animal ventilator, 24 hours after the last HDM challenge. Mice were exposed to nebulized saline (baseline measures) followed by increasing concentrations of nebulized methacholine (3–50 mg/mL) and changes in (A) Newtonian resistance (Rn), (B) tissue damping (G), (C) tissue elastance (H) were monitored. (D) Central airway sensitivity determined by calculating PC50 (amount of methacholine required to increase the baseline Rn by 50%). Statistical significance was determined for (A, B, C) using two-way repeated measures analysis of variance with Tukey’s multiple comparisons test and (D) using Mann-Whitney U test (*P≤0.05, **P≤0.01, ***P≤0.001). Asterisks in A, B, C represent comparison between HDM and HDM+IDR-1002 group. HDM, house dust mite; IDR, innate defence regulator.
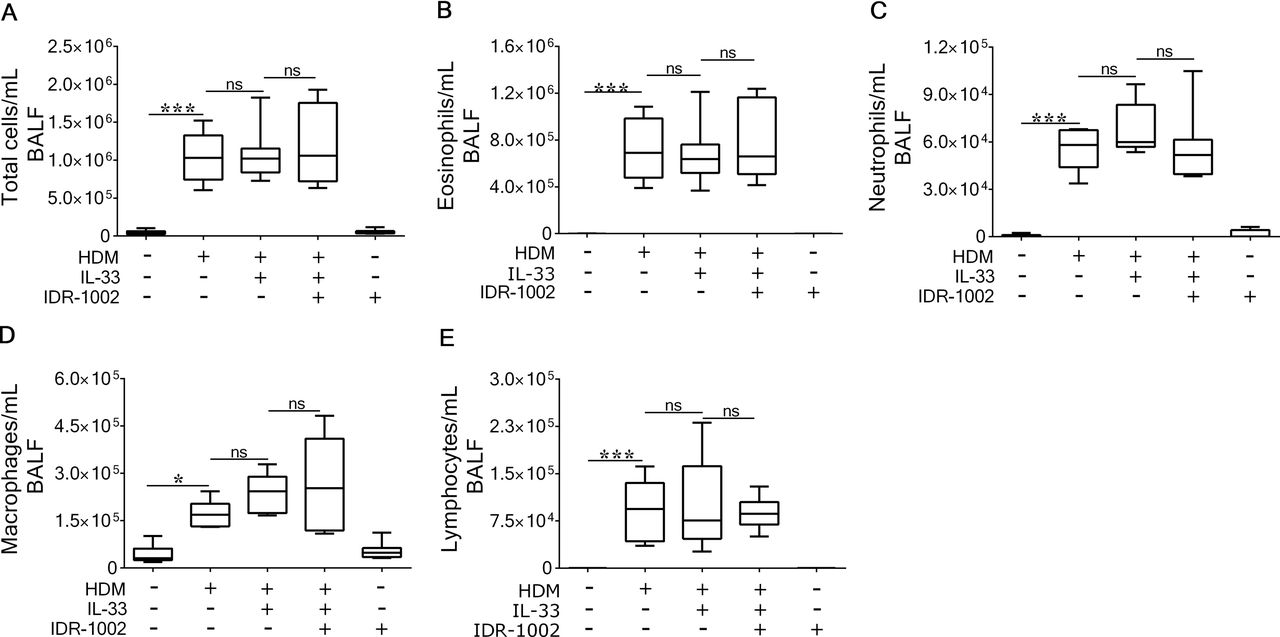
IDR-1002 suppresses leucocyte recruitment in HDM-challenged mice
We have previously shown that HDM challenge for 2 weeks induces airway inflammation that precedes airway remodelling in the murine model.29 Administration of IDR-1002 significantly reduced immune cell numbers by 35%±31% in BALF from HDM-challenged mice (figure 2A). Cell differential analyses revealed that eosinophil numbers were significantly suppressed by 51%±29% (figure 2B) and neutrophils by 29%±22% (figure 2C), whereas macrophages and lymphocytes were not affected by the peptide treatment (figure 2D and E, respectively). However, IDR-1002 treatment did not bring the cell numbers down to baseline, as immune cell numbers in BALF of HDM-challenged groups treated with IDR-1002 were higher compared with that in allergen-naive mice, and these differences were statistically significant. Administration of IDR-1002 alone did not alter total inflammatory cell numbers or differential leucocyte numbers compared with allergen-naive animals (figure 2). PAS staining of lung specimens revealed a significant increase in mucin-producing goblet cell numbers in airways of HDM-challenged mice, which was reduced by the administration IDR-1002 by ~34% (online supplementary figure 2). These findings demonstrate that administration of IDR-1002 significantly reduces infiltration of eosinophils and neutrophils in the lungs of allergen-challenged mice.

Administration of IDR-1002 significantly suppresses HDM-induced eosinophil and neutrophil infiltration in the lungs. Female BALB/c (8–10 weeks) mice were challenged with 35 µL of whole HDM extract (0.7 mg/mL) in saline intranasally for 2 weeks. IDR-1002 was administered subcutaneously three times a week at 6 mg/kg. Broncheoalveolar lavage fluid (BALF) was collected from naive (n=12), HDM-challenged (n=10), HDM+IDR-1002 (n=11) and IDR-1002 alone (n=12) mice, 24 hours after the last HDM challenge. (A) Total cell, (B) eosinophil, (C) neutrophil, (D) macrophage and (E) lymphocyte numbers were assessed. (F) Representative image of H&E staining of BALF. Bars show median and IQR, and whiskers show minimum and maximum points. Statistical significance was determined by one-way analysis of variance with Tukey’s multiple comparisons test (**P≤0.01, ***P≤0.001). HDM, house dust mite; IDR, innate defence regulator; IL, interleukin.
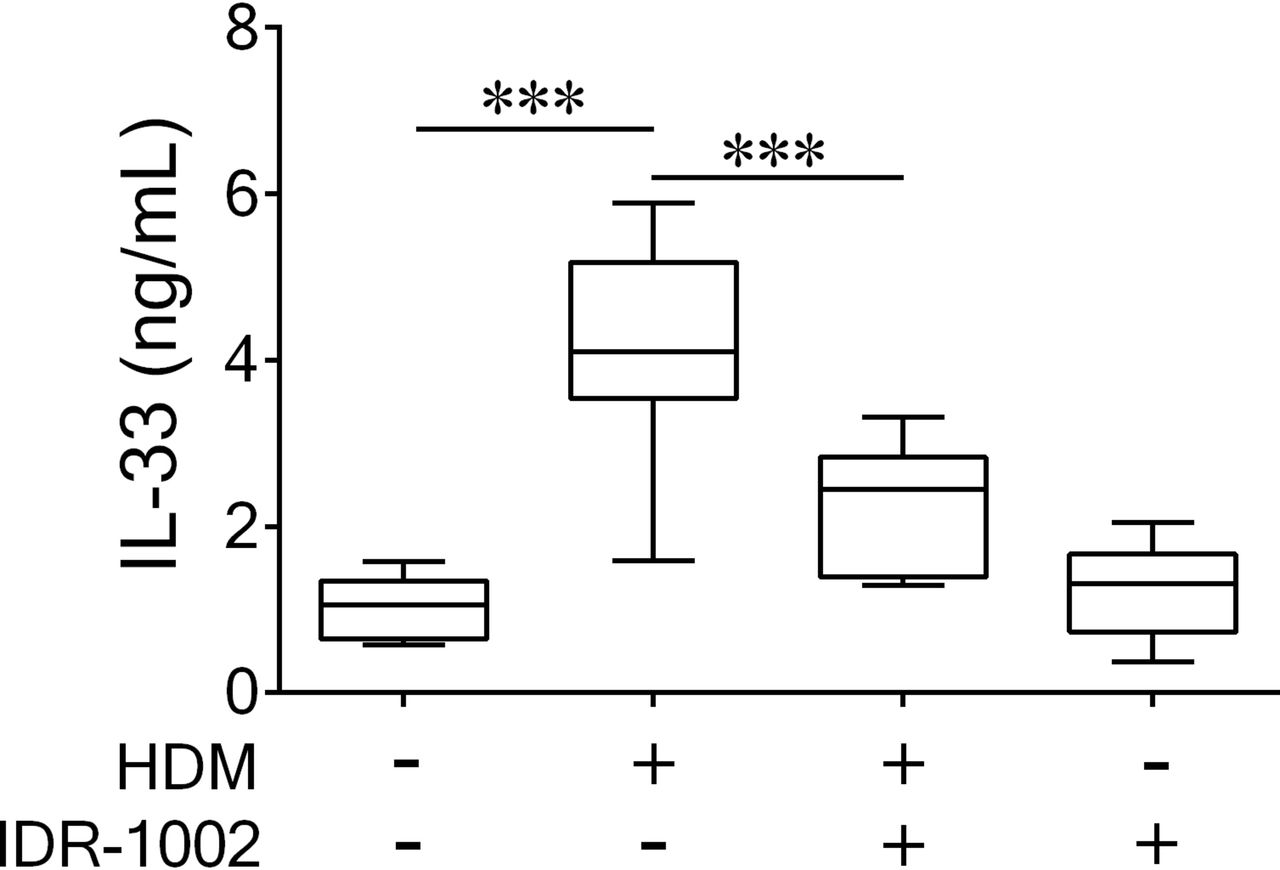
IDR-1002 suppresses production of IL-33 in HDM-challenged mice
We monitored lung tissue homogenates for the production of a panel of cytokines known to increase in this HDM model based on our previous work.29 Administration of IDR-1002 significantly suppressed the production of the proinflammatory cytokine IL-33 by 62%±30% in the HDM-challenged mice (figure 3). However, no other HDM-induced cytokine or chemokine monitored exhibited a statistically significant change in abundance following peptide administration (online supplementary figure 3). Furthermore, the peptide by itself did not significantly alter the abundance of any of the cytokines compared with naive mice at the time point monitored (online supplementary figure 3). These results suggest that IDR-1002 regulates the biosynthesis and/or secretion of the cytokine IL-33.

Administration of IDR-1002 significantly suppresses HDM-induced IL-33 in the lungs. Female BALB/c (8–10 weeks) mice were challenged with 35 µL of whole HDM extract (0.7 mg/mL) in saline intranasally for 2 weeks. IDR-1002 was administered subcutaneously three times a week at 6 mg/kg. Lung tissue was collected from naive (n=11), HDM challenged (n=11), HDM+IDR-1002 (n=9) and IDR-1002 alone (n=9) groups 24 hours after the last HDM challenge. IL-33 protein abundance was monitored in the lung tissue lysates by ELISA. Bars show median and IQR, and whiskers show minimum and maximum points. One-way analysis of variance with Tukey’s multiple comparisons test was used to assess statistical significance (***P≤0.001). HDM, house dust mite; IDR, innate defence regulator; IL, interleukin.
IDR-1002 reduces HDM+IL-33 co-challenge-induced AHR, but not leucocyte recruitment to the lungs
We have shown that IDR-1002 administration significantly suppresses IL-33 abundance in the lungs of HDM-challenged mice (figure 3). Therefore, to determine whether IDR-1002 activity is mediated by targeting IL-33, we performed a rescue experiment; recombinant murine IL-33 (1 µg/mouse) was exogenously administered intranasally on the last 5 days of HDM challenge (online supplementary figure 1B). Administration of IL-33 in the HDM-challenged mice significantly enhanced leucocyte accumulation, including that of eosinophils and neutrophils, compared with naive mice (figure 4). Administration of IDR-1002 did not reduce HDM+IL-33 co-challenge-induced accumulation of total cell numbers, eosinophils and neutrophils in the BALF (figure 4). As exogenous (additional) administration of IL-33 in HDM-challenged mice mitigated the ability of the peptide to reduce HDM-induced leucocyte accumulation in BALF, these results suggest that IDR-1002 reduces leucocyte recruitment and airway inflammation by suppressing IL-33 in the lungs of allergen-challenged mice. However, IDR-1002 maintained the ability to reduce AHR in IL-33 and HDM co-challenged mice; Rn by 73%±30%, G by 57%±24% and H by 67%±24% (figure 5), suggesting that IDR-1002 lowers AHR by mechanisms either downstream or independent of IL-33.

Exogenous administration of IL-33 mitigates the ability of the peptide to suppress immune cell infiltration to the lung of HDM-challenged mice. Female BALB/c (8–10 weeks) mice were challenged with 35 µL of whole HDM extract (0.7 mg/mL) in saline intranasally for 2 weeks, and IL-33 (1 µg per mouse) was administered intranasally on days 8–12 (online supplementary figure 1B). IDR-1002 was administered subcutaneously three times a week at 6 mg/kg. Broncheoalveolar lavage fluid (BALF) was collected from naive (n=8), HDM-challenged (n=6), HDM+IL-33 co-challenged (n=8), HDM+IL-33 co-challenged+IDR-1002 (n=7) and IDR-1002 alone (n=8) groups, 24 hours after last HDM challenge. (A) Total cells, (B) eosinophil, (C) neutrophil, (D) macrophage and (E) lymphocyte numbers were assessed in BALF. Bars shows median and IQR, and whiskers show minimum and maximum points. One-way analysis of variance with Tukey’s multiple comparisons test was used for statistical analyses (*P≤0.05, **P≤0.01, ***P≤0.001 and ns=not significant). HDM, house dust mite; IDR, innate defence regulator; IL, interleukin.

IDR-1002 reduces HDM+IL-33 co-challenge-induced airway hyper-responsiveness in mice. Female BALB/c (8–10 weeks) mice were challenged with 35 µL of whole HDM extract (0.7 mg/mL) in saline intranasally for 2 weeks, and IL-33 (1 µg per mouse) was administered intranasally on days 8–12 (online supplementary figure 1B). IDR-1002 was administered subcutaneously three times a week at 6 mg/kg. Lung mechanics was monitored using a flexiVent small animal ventilator, in naive (n=8), HDM+IL-33 co-challenged (n=8), HDM+IL-33 co-challenged +IDR-1002 (n=7) and IDR-1002 alone (n=4) groups, 24 hours after the last HDM challenge. Mice were exposed to nebulized saline (baseline measures) followed by increasing concentrations of nebulized methacholine (3–50 mg/mL), and changes in (A) Newtonian resistance (Rn), (B) tissue damping (G) and (C) tissue elastance (H) were monitored. Data shown represent the mean±SEM. Statistical significance was determined using two-way repeated measures analysis of variance with Tukey’s multiple comparisons test (*P≤0.05, **P≤0.01, ***P≤0.001). Asterisks in the figure represent comparison between HDM+IL-33 and HDM+IL-33+IDR-1002 group. HDM, house dust mite; IDR, innate defence regulator; IL, interleukin.
IDR-1002 reduces IL-33-induced AHR
We showed that IDR-1002 retained its ability to reduce AHR induced by IL-33 and HDM co-challenge in mice (figure 5). Therefore, we further examined the effects of the peptide in responses induced by IL-33 alone. IL-33 by itself induced AHR and recruitment of eosinophils and neutrophils to the lungs (figure 6A). IL-33-induced AHR was significantly blunted by IDR-1002 administration, and Rn by 56%±36%, G by 56%±23% and H by 64%±22% (figure 6A). However, the peptide did not suppress IL-33-mediated leucocyte accumulation in BALF (figure 6B). These results suggest that IDR-1002 intervenes in IL-33-induced downstream responses to reduce AHR, but not airway inflammation.

IDR-1002 reduces IL-33 alone-induced AHR, but not immune cell accumulation in the lungs. Female BALB/c (8–10 weeks) mice (n=4 for all groups) were administered (subcutaneously) with IDR-1002 (6 mg/kg) three times a week, for 2 weeks. Recombinant IL-33 (1 µg/mouse) was administered intranasally on days 8–12 (online supplementary figure 1C). Lung mechanics was monitored using a flexiVent small animal ventilator, 24 hours after the last IL-33 challenge. Mice were exposed to nebulized saline (baseline measures) followed by increasing concentrations of nebulized methacholine (3–50 mg/mL), and changes in (A) Newtonian resistance (Rn), tissue damping (G) and tissue elastance (H) were monitored. Data shown represent the mean±SEM. Statistical significance was determined using two-way repeated measures analysis of variance (ANOVA) with Tukey’s multiple comparisons test. Asterisks in the figure represent comparison between IL-33 and IL-33 +IDR-1002 groups. (B) Broncheoalveolar lavage fluid (BALF) was collected 24 hours after the last IL-33 challenge. Total cells, eosinophil, neutrophil, macrophage and lymphocyte cell numbers were assessed in BALF. Bars show median and IQR, and whiskers show minimum and maximum points. One-way ANOVA with Tukey’s multiple comparisons test was used for statistical analyses (*P≤0.05, **P≤0.01, ***P≤0.001 and ns=not significant). AHR, airway hyper-responsiveness; IDR, innate defence regulator; IL, interleukin.
IDR-1002 abrogates IL-33 production in human PBECs
We further validated the activity of IDR-1002 in mitigating the production of IL-33 in human PBECs. Previous studies have shown that a cytomix of IFN-γ and TNF-α induces the production of IL-33.35–37 Therefore, we stimulated human PBECs isolated from four independent human donors (n=4), with recombinant human IFN-γ (30 ng/mL) and/or TNF-α (20 ng/mL), in the presence and absence of IDR-1002 (20 or 40 µM). The concentration range of IDR-1002 was selected from previous in vitro studies.38 IL-33 abundance was significantly increased in response to either IFN-γ or the cytomix (IFN-γ+TNF-α), but not TNF-α alone (figure 7A,B), suggesting that IFN-γ drives IL-33 production in human PBECs. IDR-1002 abrogated cytomix-induced IL-33 protein abundance in human PBECs (figure 7A,B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IDR-1002 significantly reduces IL-33 production in human primary bronchial epithelial cells (PBECs). Human PBECs obtained from four independent donors (n=4) were stimulated with TNF-α (20 ng/mL) and/or IFN-γ (30 ng/mL), in the presence and absence of IDR-1002 (20 and 40 µM). Cytoplasmic fraction of cells isolated 24 hours poststimulation was probed in immunoblots to assess the abundance of IL-33, IRF1 and IP10. Protein abundance was quantified by densitometry. (A) A representative immunoblot, and densitometry analysis for (B) IL-33, (C) IRF1 and (D) IP10 is shown. Y-axis represents relative band intensity compared with that in unstimulated cells normalised to 1, after normalisation with β-actin for protein input. Each dot represents an individual donor, and bars show the median and IQR. Repeated measures one-way analysis of variance with Fisher’s least significant difference test was used for statistical analyses (*P≤0.05, **P≤0.01). IDR, innate defence regulator; IFN-γ, interferon gamma; IL, interleukin; IP10, inducible protein 10; IRF1, interferon response factor 1; TNF-α, tumour necrosis factor alpha.
To further evaluate the specificity of IDR-1002 activity, we repeated these experiments using a related peptide IDR-1 (KSRIVPAIPVSLL-NH2). Similar to IDR-1002, IDR-1 is also derived from the bovine cathelicidin Bac2A. However, in contrast to IDR-1002, IDR-1 does not suppress inflammatory cytokine-mediated signalling and downstream responses in human cells.38 Peptide IDR-1 did not suppress IL-33 production in PBECs (online supplementary figure 4).
As IL-33 production was primarily driven by IFN-γ-mediated response (figure 7A and B), we further examined the effect of IDR-1002 on expression of response elements induced by the IFN-γ canonical signalling, namely transcription factor interferon response factor 1 (IRF1) and downstream IFN-inducible protein 10 (IP10).39 Production of IFN-γ-induced IP10 and IRF1 was not suppressed by IDR-1002 (figure 7C and D, respectively).
Discussion
In this study, we demonstrate that an immunomodulatory IDR peptide, IDR-1002, reduces AHR, leucocyte recruitment (eosinophils and neutrophils) to the lungs and prevents epithelial goblet cell hyperplasia, which is otherwise induced by repeated allergen challenge in a murine model of allergic asthma. We demonstrate that the peptide suppresses cytokine IL-33 production in murine lungs and in human PBECs. We show that the ability of the peptide to suppress accumulation of eosinophils and neutrophils in the lungs of HDM sensitised and challenged mice is abrogated by additional administration of the cytokine IL-33, thus suggesting that suppression of IL-33 production is essential for IDR-1002 to reduce airway inflammation. We also show that IDR-1002 exhibits the ability to reduce AHR induced by either HDM, IL-33 or HDM+IL-33 co-challenge in vivo. Taken together, our findings suggest that the peptide engages different mechanisms that do not completely overlap to control AHR and airway inflammation. Overall, in this study we demonstrate the potential of a synthetic immunomodulatory IDR peptide to alleviate airway inflammation and hyper-responsiveness, and intervene in the pathobiology of allergic asthma.
IDR peptides have been demonstrated to modulate immune responses to control infections and limit excessive inflammation in various models.20 21 38 40–45 Some IDR peptides are currently in phase II/III clinical trials primarily for infectious diseases.46–48 The use of IDR peptides for chronic inflammatory diseases such as asthma has not yet been fully explored. We have previously demonstrated that IDR-1002 suppresses cytokine-mediated inflammation in human stromal cells in vitro.38 Similarly, a recent study has shown that IDR-1002 attenuates inflammation in cystic fibrosis airway cells in vitro.23 This is the first study to demonstrate the beneficial use of a synthetic IDR peptide in an in vivo HDM-challenge model of allergic asthma. A recent study demonstrated that administration of natural cathelicidin HDPs such as the human LL-37 and murine cathelin-related antimicrobial peptide (CRAMP) leads to an increased inflammatory response in ovalbumin-challenged mice.49 A critical distinction of our study is that we use a synthetic IDR peptide that is designed to optimise immunomodulatory functions without the associated cytotoxic and proinflammatory effects of natural HDPs at mucosal surfaces.43 Synthetic IDR peptides selectively reduce exacerbated inflammatory responses while maintaining the natural functions of endogenous HDPs to resolve infections.43 47 The increased inflammatory response reported by Jiao et al 49 may also be due to the mode of peptide delivery, as they administered cathelicidin HDPs intranasally, whereas, we employed subcutaneous administration of the synthetic IDR peptide in this study. It is thus possible that the modulation of inflammatory responses in the lung may be dependent on the route of delivery of immunomodulatory peptides.
In this study, we demonstrate that peptide IDR-1002 abrogates IL-33 production in a murine model of allergic asthma and in human PBECs, and inhibits IL-33-induced AHR in vivo. The cytokine IL-33 is a member of the IL-1 family of cytokines and acts as an early cytokine inducer of allergic airway inflammation and asthma by promoting eosinophil and neutrophil recruitment and activation.50 The IL-33/ST2 receptor axis plays a central role in mediating Th2-biased and glucocorticoid-resistant airway inflammation.51 52 Recent studies show that IL-33 is significantly increased in severe asthmatics and is a key mediator of steroid-refractory responses in humans.25–27 In line with this, studies in multiple animal models demonstrate that corticosteroid therapy has no effect on IL-33 lung levels.25 28 Consequently, IL-33 is an attractive therapeutic target for chronic airway inflammatory diseases such as severe asthma. Therefore, our findings on the effects of the IDR peptide on IL-33 production and action, and in the processes of allergic airway inflammation and AHR, may contribute to the development of novel therapies for steroid-refractory severe asthma.
IL-33 is known to increase the expression of the KC receptor CXCR2, which can regulate migration of neutrophils.53 However, the role of IL-33 to increase neutrophil migration is only partially dependent on the KC-CXCR2 axis,54 as IL-33 by itself is also capable of directly increasing neutrophil migration.49 Consistent with this, we demonstrate that IL-33 alone increases leucocyte accumulation, including neutrophils, in the lungs. This suggests that IL-33 may play a role as a driver of neutrophilia in the HDM-challenged murine model. As IDR-1002 did not suppress HDM-induced KC expression in the lungs, the peptide may reduce HDM-induced neutrophil lung influx by suppressing IL-33 production. This is supported by our results demonstrating that exogenous administration of IL-33 mitigates the capacity of the peptide to blunt HDM-induced neutrophil recruitment to the lungs and that IL-33 alone-induced neutrophil accumulation in BALF is not reduced by the peptide. Taken together, these results suggest that the suppression of IL-33 production is an essential underpinning for the ability of the peptide to control leucocyte recruitment to the lungs.
Despite our observations that IDR-1002 prevents goblet cell hyperplasia that can be promoted by IL-5 and IL-13, we did not observe any effects of IDR-1002 on IL-5 or IL-13 production in response to allergen challenge in the murine model. This suggests that the peptide may prevent goblet cell hyperplasia by engaging alternate mechanisms or that it alters the kinetics of responses to IL-5 or IL-13, and thus may not be captured at the time point we measured outcomes in this study. Nevertheless, we have established the feasibility of exploring an IDR peptide-based immunomodulatory therapy for reducing airway inflammation in allergic asthma, the core mechanism of which appears to be linked to pathways that regulate IL-33 production.
IL-33 is expressed by human epithelial cells,25 28 and acute inflammatory cytokines TNF-α and IFN-γ can induce IL-33 production in epithelial cells.35 36 Recent studies corroborate an association of TNF-α and IFN-γ with severe steroid-resistant asthma.26 55 Aligned with these studies, we demonstrate a significant increase in IL-33 production in response to cytomix (TNF-α+IFN-γ) in human PBECs, and show that this is primarily driven by IFN-γ. We conclusively demonstrate that the peptide IDR-1002 abrogates IL-33 abundance that is enhanced in response to IFN-γ and TNF-α cytomix in human PBECs. Interestingly, our mechanistic studies show that IDR-1002 does not mitigate response elements of the IFN-γ-induced pathway such as IRF1 or IP10, suggesting that the peptide targets signalling intermediates independent of the IFN-γ-induced canonical pathway to suppress IL-33 production. Considering that the IFN-γ-mediated canonical pathway plays a major role in the ability to fight infections,56 and as IDR peptides are known to protect against infections,20–22 40 it is not surprising that IDR-1002 activity does not intervene in the IFNγ-canonical pathway. Unfortunately, upstream pathways responsible for the production of IL-33 are not well known. Therefore, future studies using broad omics-based approaches will be required to delineate the full repertoire of signalling mechanisms that underlie the ability of IDR-1002 to suppress IFN-γ-induced IL-33 production, which is beyond the scope of this manuscript.
As airway inflammation is often correlated with AHR,25 we initially speculated that the suppression of IL-33 by IDR-1002 might be essential for preventing allergen-induced lung dysfunction. However, we show that the protective effects of IDR-1002 on allergen-induced AHR are maintained even after exogenous (additional) administration of IL-33. Therefore, we examined whether the peptide intervenes in AHR and/or airway inflammation induced by IL-33 alone challenge. We show conclusively that IL-33-induced AHR, but not airway inflammation, is significantly reduced by IDR-1002. These results suggest that the ability of IDR-1002 to reduce AHR is not only explained by a reduction of IL-33 production but also involves mechanisms downstream or independent from IL-33. Our findings align with recent studies using multiple antigens in mouse models that show airway inflammation and AHR are mediated by a complex network of biological pathways which do not fully overlap.57 58 Furthermore, in humans there are disparate cause–effect relationships between airway inflammation and AHR.59 60 Taken together, our results suggest that the activity of IDR-1002 to mitigate airway inflammation and AHR likely engages different signalling pathways and offers the opportunity to use IDR peptides in future studies as a probe to delineate unique molecular mechanisms that link airway inflammation and AHR.
A distinct advantage of developing an IDR peptide-based immunomodulatory therapy for asthma is the additional benefit of these peptides in resolving infections. IDR peptides can control pulmonary infections including multidrug-resistant TB and biofilm infections recalcitrant to antibiotics.21 22 Specifically, IDR-1002 protects against multiple bacterial infections including TB and exhibits potent antibiofilm activity against Pseudomonas aeruginosa, which is a major problem in chronic pulmonary disease.20 21 24 Recent studies have demonstrated that IL-33 is a key mechanistic link in viral infection-induced synergistic exacerbation of allergic asthma.61 This is consistent with previous reports demonstrating that patients with severe asthma are more susceptible to pulmonary infections.62 Therefore, our finding on the ability of IDR-1002 to alleviate allergic airway inflammation and AHR, taken together with previous studies demonstrating its potent antibacterial and antibiofilm activity, suggests that IDR-1002-based new therapies may be beneficial for asthma without compromising the patients’ ability to resolve infections.
In conclusion, in this study we have established the therapeutic potential of an immunomodulatory IDR peptide, IDR-1002, in allergic asthma. The mechanisms underlying the activity of IDR-1002 involve suppression of the steroid-resistant mediator IL-33, thus suggesting the potential of IDR-1002-based development of new therapies to be beneficial for severe asthma. This study provides the foundation to develop IDR peptides such as IDR-1002 as an immunomodulatory therapy with the dual potential to control chronic airway inflammatory diseases and infections.
Acknowledgments
The authors gratefully acknowledge technical assistance of Dennis Ninaber at University of Leiden and Protiti Khan at University of Manitoba for this study.
References
Footnotes
Contributors HP performed majority of the experiments and data analyses, contributed to the development of the scientific concepts and wrote the manuscript. MH and AA performed various assays for monitoring endpoint responses and cytokine production in the animal tissues. SB performed the murine lung function analyses. AJH provided significant intellectual input in the development of this study and extensively edited the manuscript. AMvdD provided extensive intellectual support in the design of the experiments using human primary cells. PSH provided significant intellectual input and directly supervised the experiments using primary human cells. NM conceived and directly supervised the study and extensively edited the manuscript. All authors reviewed and edited the manuscript.
Funding Funding support for this study was obtained from the Manitoba Health Research Council (MHRC); The Dr Paul T. Thorlakson Foundation, Winnipeg, MB, Canada; the Canadian Respiratory Research Network (CRRN) and the Children’s Hospital Research Institute of Manitoba (CHRIM). HP and AA were supported by studentships from MHRC.
Competing interests NM and HP are inventors on a patent application filed with Canadian Intellectual Property Office (WO2015077888) that incorporates aspects of the findings described in this manuscript. Other authors have no competing interest to declare.
Patient consent Not required.
Ethics approval University of Manitoba Animal Care Ethics Board.
Provenance and peer review Not commissioned; externally peer reviewed.