Article Text

Statistics from Altmetric.com
Marie Wright (Specialist Registrar): A 4-month-old female infant was referred with a 4-week history of tachypnoea and faltering growth. She was the first child of healthy unrelated parents, and was conceived following two early miscarriages. Noteworthy family history included the unexplained sudden deaths of her mother’s sibling aged 8 months and a 12-year-old first cousin. The pregnancy was complicated by intrauterine growth restriction, and birth weight at 37 weeks’ gestation was 2.5 kg (second centile). She had no respiratory compromise in the neonatal period, and initially established good weight gain and met developmental milestones age appropriately. At 12 weeks old, she was observed by her parents to be tachypnoeic. Her work of breathing gradually increased over the next month, but was not associated with cough, stridor or systemic symptoms. Simultaneously, her weight drifted to below the 0.4th centile.
At first presentation, the infant appeared malnourished but was alert and interactive. She had marked respiratory distress and a paradoxical breathing pattern with indrawing of the abdomen on inspiration, but respiratory examination was otherwise unremarkable. Muscle strength and tone were normal, but she had positional talipes and correctable flexion deformities of her fingers.
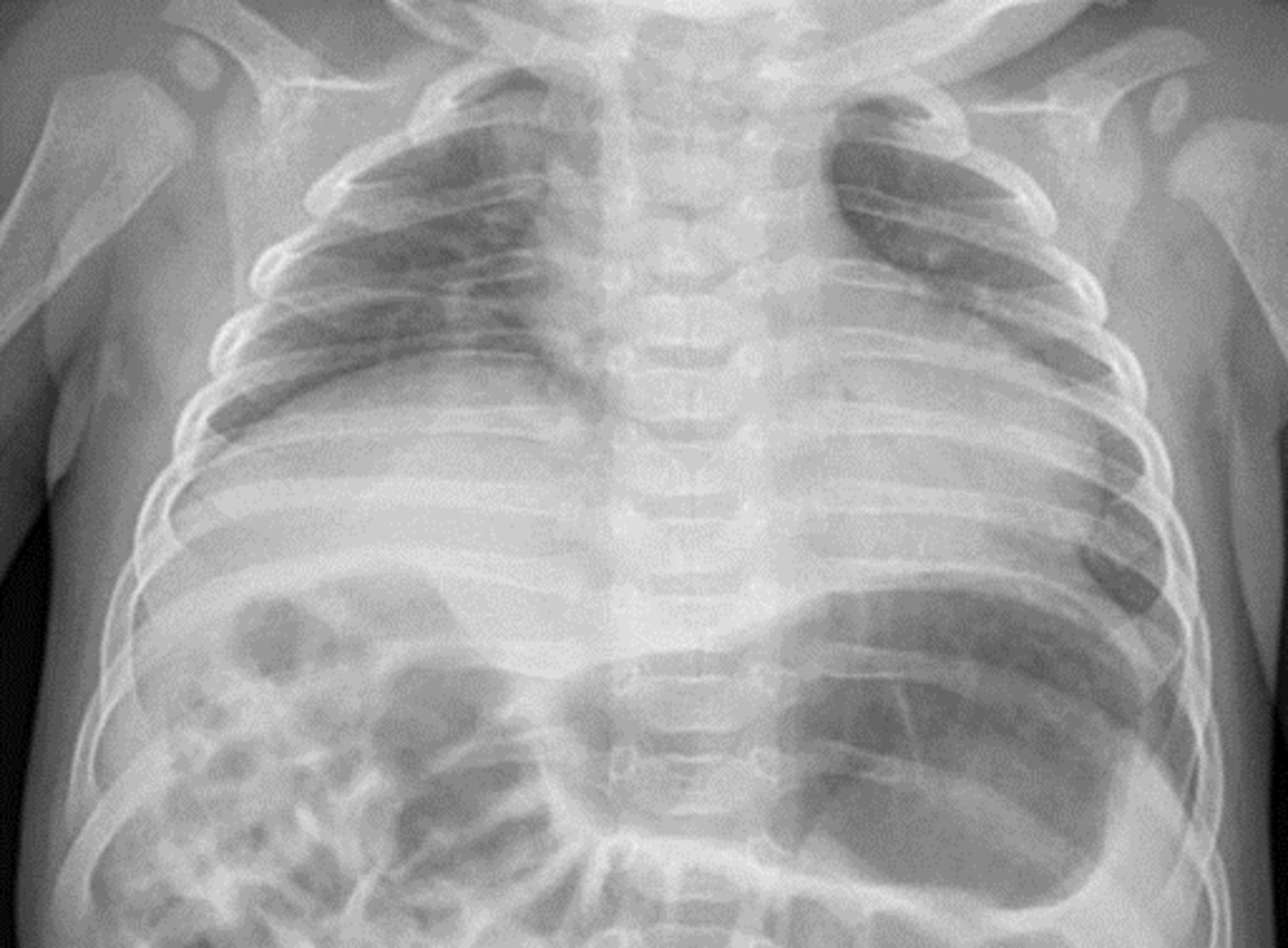
Oxygen saturation was 95% in air, and gas exchange was normal on capillary blood gas analysis (pH 7.44, pCO2 4.66 kPa, HCO3 24 mmol/L, BE −0.2 mmol/L). ECG and echocardiogram were normal. Chest X-ray showed an elevated right hemidiaphragm (figure 1), and unilateral diaphragmatic paralysis was confirmed on fluoroscopic ultrasound. Rhinovirus and Adenovirus were detected on nasopharyngeal aspirate viral PCR.

{kind=link}
Chest X-ray at 4 months of age.
Andy Bush (Professor of Respiratory Paediatrics): These clinical features are suggestive of a progressive neuromuscular disorder with relative preservation of limb muscle function and early respiratory muscle involvement. There are several possible causes; the likeliest diagnosis is spinal muscular atrophy with respiratory distress type 1 (SMARD1), and the differential diagnosis includes nemaline rod myopathy, subtypes of congenital muscular dystrophy, and congenital myopathies including MEGF10 gene-related early-onset myopathy, areflexia, respiratory distress and dysphagia. SMARD1 is a neuromuscular disorder characterised by early diaphragm weakness and is caused by mutations of the IGHMBP2 gene on chromosome 11q13.3. Despite their similar names, SMARD1 is genetically unrelated to chromosome 5q-linked spinal muscular atrophy (SMA) which presents as a markedly floppy infant with intercostal muscle weakness but relative sparing of the diaphragm, and has a characteristic respiratory pattern with chest wall indrawing and exaggerated abdominal rise on inspiration.
SMARD1-affected infants typically present between 6 weeks and 6 months of age with respiratory distress secondary to diaphragmatic weakness. In retrospect, there is a preceding history of intrauterine growth restriction, reduced fetal movements or preterm delivery in many instances. Respiratory insufficiency commonly presents with the child’s first viral respiratory tract infection, as in this case, or as an unexplained acute life-threatening event. Diaphragmatic paralysis is initially unilateral, giving the classic appearance of a raised hemidiaphragm on chest X-ray. In the context of rapidly progressive respiratory insufficiency, this appearance should raise suspicion of SMARD1. The initial priorities for this child are to stabilise her respiratory function and arrange an urgent neurological review to get a precise diagnosis.
MW: Within a week of admission, her respiratory distress had worsened and serial blood gas analysis showed evolving respiratory failure. Non-invasive ventilation (NIV; pressures 18/6, rate 35, ResMed Stellar 3000 ventilator in S/T mode) was initiated, and her work of breathing subsequently diminished and gas exchange normalised. However, from the outset she was dependent on NIV and could not tolerate even brief periods off support. By 5 months of age, her peripheral muscle strength had deteriorated with limited residual antigravity movement of the fingers and ankles. She was referred for neurological assessment.
Adnan Manzur (Consultant Paediatric Neurologist): This clinical course is typical of SMARD1. Irreversible degeneration of α-motor neurons (anterior horn cells) in the spinal cord leads to rapidly progressive respiratory failure followed by distal muscle weakness and wasting. Ultimately, a flaccid paralysis develops with areflexia and loss of antigravity movement in the lower limbs. Autonomic dysfunction is also common and can lead to tachycardia, excessive sweating, urinary retention and constipation.
Longitudinal studies of SMARD1 describe that affected children are rarely able to sit without support and can never walk, although in some cases a plateau in neuromuscular decline is seen after 1 year of age.1 Permanent respiratory support is invariably needed, without which survival rarely exceeds the first year of life.2 Long-term survival has been described with provision of NIV or mechanical ventilation via tracheostomy, although whether these interventions are in the best interests of the child is a contentious matter.3
SMARD1 is definitively diagnosed by genetic analysis, but results can take many weeks or months by which point the child may have developed respiratory failure necessitating mechanical ventilation. Electrophysiological tests including nerve conduction studies (NCS) and electromyography (EMG) provide a more rapid result, and can be used in cases of clinical suspicion to support the diagnosis before genetic results are available. In this case, NCS showed abnormally small compound muscle action potential amplitudes on motor stimulation in conjunction with normal sensory studies, and EMG demonstrated neurogenic changes. These abnormalities are characteristic of anterior horn cell dysfunction and compatible with a diagnosis of SMARD1.
Respiratory failure with primary diaphragmatic weakness and progressive distal muscle weakness are useful clinical clues indicating the need to prioritise genetic tests for SMARD1, particularly when supported by compatible investigation findings. It is important to clearly differentiate SMARD1 from the far more common chromosome 5q-linked SMA, which has a very different paradoxical pattern of breathing with relative sparing of the diaphragm, proximal more than distal weakness and is responsive to early administration of novel intrathecal nusinersen treatment.
MW: While the results of diagnostic testing were awaited, the child’s condition improved dramatically; she was alert, splashing happily in her bath and interacting well with her parents, although she remained completely reliant on NIV. Her parents wished to wait for the genetic results before discussing further management.
AB: Sequencing of the IGHMBP2 gene identified compound heterozygous mutations. Confirmation of the SMARD1 diagnosis and its terrible prognosis were a devastating blow to the parents. We had an open and realistic discussion about their child’s prognosis and her treatment options, which were to withdraw treatment altogether, provide continuous NIV or perform a tracheostomy. The family opted to take her home on continuous NIV and with a feeding gastrostomy, and she was discharged home at 8 months of age.
MW: On subsequent outpatient review up to her current age of 18 months, her ventilatory requirements have remained unchanged despite an excellent growth trajectory (weight 9th–25th centiles, height 25th centile). Remarkably, by 12 months of age she could crawl and pull to stand, and by 15 months she was walking with support. Orthoses had been fitted to minimise any functional limitation caused by ankle and finger contractures. She was vocalising appropriately for age and managing oral secretions without the need for suction or a ‘cough assist’ device.
AB: Clearly this child’s progress came as a complete surprise to everyone. We had a further discussion with the family about options for respiratory support. They now wished for a tracheostomy given her developmental progress, and this was inserted at age 18 months.
This case illustrates two important points. The first is that in an era of support of inspiratory and expiratory muscle function, and enteral nutrition, the prognosis for neuromuscular diseases has changed dramatically, and it is unwise to be too dogmatic based on old data.4 Without nutritional and respiratory support, this baby would undoubtedly have died quickly, but her case highlights that some children with SMARD1 have the potential to defy expectations and achieve a quality of life that may justify more invasive management approaches like tracheostomy or gastrostomy insertion. Other children with milder phenotypes have been described, including rare exceptions who have survived into adulthood and need respiratory support only at night. Siblings with an identical mutation but strikingly different clinical outcomes have also been described,5 suggesting that there is potential for modifier loci to alter the phenotypic expression of the disease.
The second point is the need to re-evaluate the ethics of treatment options as the clinical picture evolves. The child’s parents were always clear that her quality of life was paramount, and initially felt that tracheostomy insertion was unjustified given her poor prognosis. However, this was re-evaluated considering her unexpectedly good developmental trajectory, and tracheostomy was subsequently felt to offer a better quality of life than continuous NIV. Tracheostomy can benefit children with neuromuscular diseases who are clinically stable but NIV dependent by increasing their potential for neurodevelopmental progress and preventing sequelae of continuous mask use including skin irritation and midfacial deformity. However, the potential benefits must be balanced against the risk of subsequent physical decline which could leave the child in a permanent ‘locked in’ state following tracheostomy insertion.3
Although the prognosis of neuromuscular diseases may be unpredictable, it is essential to open discussions with parents about ceilings of care soon after the diagnosis is suspected. In every case of early-onset respiratory muscle failure, irrespective of clinical severity at the time of diagnosis, the dignity and best interests of the child must be of paramount importance. Early confirmation of the diagnosis, maintaining an open and honest dialogue with the parents, considering each case on an individual basis and being ready to reverse previous decisions as the child progresses are key steps towards achieving this.
Footnotes
Contributors MW and AB conceptualised the paper and wrote the first draft. AM edited the manuscript and provided expert insight. All authors have read and approved the final draft of the manuscript.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.