Article Text

Abstract
Background Elevated plasma levels of angiopoietin-2 (ANGPT2) have been reported in patients with acute lung injury (ALI); however, it remains unclear whether this increase contributes to, or just marks, the underlying vasculopathic inflammation and leak associated with ALI. Here we investigated the biological consequences of inducing high circulating levels of ANGPT2 in a mouse model of endotoxin-induced ALI.
Methods Transgenic mice (ANGPT2OVR) with elevated circulating levels of ANGPT2, achieved through conditional hepatocyte-specific overexpression, were examined from 3 to 72 hours following lipopolysaccharide (LPS)-induced ALI. An aptamer-based inhibitor was used to neutralise the effects of circulating ANGPT2 in LPS-exposed ANGPT2OVR mice.
Results Total cells, neutrophils and macrophages, as well as inflammatory cytokines, were significantly higher in bronchoalveolar lavage (BAL) of ANGPT2OVR versus littermate controltTA mice at 48 hours and 6 hours post-LPS, respectively. In contrast, LPS-induced vascular leak, evidenced by total BAL protein levels and lung wet/dry ratio, was unchanged between ANGPT2OVR and controlstTA, while BAL levels of IgM and albumin were decreased in ANGPT2OVR mice between 24 hours and 48 hours suggesting a partial attenuation of vascular leak. There was no significant difference in LPS-induced mortality between ANGPT2OVR and controlstTA. An ANGPT2-neutralising aptamer partially attenuated alveolar cell infiltration while exacerbating vascular leak in LPS-exposed ANGPT2OVR mice, supported by underlying time-dependent changes in the lung transcriptional profiles of multiple genes linked to neutrophil recruitment/adhesion and endothelial integrity.
Conclusions Our findings suggest that high circulating ANGPT2 potentiates endotoxin-induced lung inflammation but may also exert other pleiotropic effects to help fine-tune the vascular response to lung injury.
- Ards
- bacterial Infection
- cytokine biology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
Elevated plasma levels of angiopoietin-2 (ANGPT2) in acute lung injury (ALI)/acute respiratory distress syndrome patients are associated with poor prognosis; however, it remains unclear whether these elevated circulating levels are just a marker or mediator of underlying pulmonary vascular dysfunction.
What is the bottom line?
For the first time, this study demonstrates that elevated plasma levels of ANGPT2 play a context-dependent role in fine-tuning the inflammatory response to ALI.
Why read on?
The isolated elevation of circulating ANGPT2 via hepatocyte-specific overexpression and secretion exacerbated pulmonary inflammation, but not vascular leak, and did not alter mortality in a murine model of bacterial endotoxin-induced ALI.
Introduction
Acute respiratory distress syndrome (ARDS) is a life-threatening condition for which there is currently no specific pharmacological treatment.1 Pulmonary inflammation and vascular leak are hallmarks of acute lung injury (ALI) seen in ARDS, which may be mediated in part through dysregulation of the angiopoietin–Tie2 signalling axis.2 Angiopoietin-1 (ANGPT1) and angiopoietin-2 (ANGPT2) are ligands for the endothelial cell (EC)-enriched TIE2 receptor tyrosine kinase. Paracrine release of ANGPT1 from pericytes promotes TIE2 receptor phosphorylation and downstream activation of signalling pathways that protect against inflammation3 4 and vascular leak.5–7 In contrast, ANGPT2 (stored in Weibel-Palade bodies of ECs) has been shown to competitively inhibit ANGPT1-induced TIE2 phosphorylation in vitro, and its transgenic overexpression in the mouse embryo has been reported to cause defects in vascular development mimicking those seen in ANGPT1 and TIE2 knockout animals.8–10 While these early observations support the classic view of ANGPT2 as a natural TIE2 antagonist and vascular destabilising factor,9 there is increasing evidence that ANGPT2 may also function as a TIE2 agonist and vasculoprotective factor in a context-dependent manner. A number of in vitro studies have shown that ANGPT2 can activate the TIE2 receptor under various circumstances including in non-ECs lacking inhibitory coreceptors,11 over extended periods of time12 or at very high concentrations.13 14 In vivo studies in mice have also shown that ANGPT2 can inhibit vascular leak induced by inflammatory stimuli such as mustard oil.15 Thus, the role of ANGPT2 may vary widely from vasculopathic to vasculoprotective factor depending on the specific biological context.
In clinical settings, plasma levels of ANGPT2 have been shown to be elevated in patients with inflammatory disorders, in particular ALI/ARDS and sepsis,16–20 and in some cases were associated with the severity of lung injury21 and mortality.17 However, while these observational studies establish the clinical relevance of ANGPT2 and suggest a potential role as a prognostic biomarker, they provide limited information on whether the elevated circulating levels contribute causally to underlying disease activity in ALI. To date, and to the best of our knowledge, no study has directly interrogated the potential contributory role of high circulating levels of ANGPT2 in an animal model of ALI.
Here, we hypothesised that endotoxin-induced ALI would be exacerbated by systemic ANGPT2 overexpression. High plasma levels of ANGPT2 were induced in transgenic mice (ANGPT2OVR) independent of lung injury, via conditional hepatocyte-driven overexpression and secretion, and the effects on pulmonary vascular inflammation, permeability and mortality were then evaluated in the lipopolysaccharide (LPS)-induced model of ALI. Neutralisation of the circulating ANGPT2 in ANGPT2OVR mice, via aptamer-based inhibition, was used to further probe the potential causal role of ANGPT2 on pulmonary inflammation and leak and identify molecular pathways involved in ANGPT2-mediated pulmonary transcriptional reprogramming.
Materials and methods
Peripheral blood samples from healthy subjects were obtained with informed written consent between years 2011 and 2014 at a single centre, in accordance with protocols and regulations approved by the Ottawa Hospital Research Ethics Board (#2011470–01H). Peripheral blood samples from patients with ALI/ARDS secondary to septic shock were obtained with informed written consent in 2009 in accordance with protocols and regulations approved by the respective research ethics boards in the multicentre PRECISE pilot trial.22 All animal procedures were approved by the University of Ottawa’s Animal Care Ethics Committee and complied with the principles and guidelines of the Canadian Council on Animal Care. All animal, cellular and molecular experiments and related data analysis were conducted over a period of 7 years from 2010 to 2017.
Transgenic overexpression of ANGPT2
High systemic circulating levels of human ANGPT2 (hANGPT2) were achieved with a doxycycline (Dox)-based conditional transgenic mouse system as described previously.23 Of note, transgenic overexpression of ANGPT2 in mouse embryos has previously been shown to lead to midgestational embryonic lethality.9 In the conditional system used in the current study, the tetracycline transactivator (tTA; driver transgene) expressed from a liver-specific promoter binds to tetracycline operator sequences upstream of the ANGPT2 responder transgene. In the presence of the tetracycline analogue, Dox (Harlan chow), the expression of ANGPT2 is blocked. In this conditional system, the expression of ANGPT2 was induced by the dietary withdrawal of Dox. All mice were taken off Dox at 3 weeks and used for experiments at 8–10 weeks of age. Double transgenic ANGPT2OVR mice (containing both the tTA driver gene and ANGPT2 responder gene) and single transgenic littermate controltTA mice (containing just the tTA driver gene) were confirmed by PCR genotyping using DNA extracted from ear biopsies and transgene-specific primers as described previously.23 Mice were on a CD1 genetic strain background. ANGPT2OVR mice were indistinguishable in size and appearance from littermate controls and represented approximately 25% of each litter.
LPS-induced model of ALI
Mice received an intratracheal instillation of LPS (Escherichia coli 055:B5; 1 mg/kg or 500 000 EU/kg body weight in normal saline; Sigma), and blood, bronchoalveolar lavage (BAL) and lungs were collected after 3 hours, 6 hours, 24 hours, 48 hours and 72 hours. Of note, data for each time point represent a separate set of mice rather than serial measurements from a single set of mice. An LPS dose of 10 mg/kg was used in experiments to assess LPS-induced mortality.
Anti-ANGPT2 aptamer treatment in ANGPT2OVR mice
The anti-ANGPT2 RNA aptamer, which binds to hANGPT2 protein, was developed previously.24 The aptamer and a scrambled sequence control RNA were synthesised by Samchully Pharmaceutical (Seoul, Korea), with 2′-fluoropyrimidines and terminal 3′−3′ inverted deoxythymidine caps for nuclease resistance. The non-functional control RNA was composed of the same number and type of chemically modified nucleotides, but the sequence of nucleotides was randomly scrambled. Aptamer or scrambled-sequence control RNA (0.1 mg/kg, dissolved in phosphate buffered saline (PBS)) were injected in ANGPT2OVR mice via tail vein injection by an operator that was blinded to the treatment status. No transfection agent was used with the aptamer or scrambled control RNA to minimise the potential for cellular uptake. All mice were then given LPS (1 mg/kg) via intratracheal instillation (within 5 min). Mice were euthanised for BAL analysis after 48 hours.
Biochemical measurements
Total protein levels in BAL were measured by bicinchoninic acid (BCA) assay. IgM and albumin levels in BAL were measured by ELISA (IgM, cat# E90-101; albumin, cat# E90-134, Bethyl laboratories) according to manufacturer instructions. ANGPT and TIE2 protein levels were measured by ELISA (hANGPT2, cat# DANG20, R&D Systems; mouse ANGPT2, cat# MANG20, R&D Systems; mouse ANGPT1, cat# EK1296, Boster Biological Tech Co. Ltd.; mouse total TIE2, cat #DYC762, R&D Systems; mouse phospho-TIE2 (p-TIE2), cat# DYC2816, R&D Systems) according to manufacturer instructions. BAL cytokine levels (cat# MAGPMAG-24K, Millipore) and phospho Akt/total Akt levels (cat# 48-618MAG, Millipore) in mouse lung homogenates were measured by multiplex magnetic bead-based luminex kits and run on a Bioplex 200 system (Biorad).
Statistics
The unpaired two-sided Student’s t-test (for parametric data) or Mann-Whitney U test (for non-parametric data) were used for two-group comparisons as appropriate. Gaussian distributions were assessed using the D’Agostino Pearson omnibus normality test. For three or more group comparisons, one-way or two-way analysis of variance was performed with post hoc tests for individual p values with or without adjustment for multiple comparisons as reported in each figure legend. Gene expression profiling was corrected for multiple comparisons using the false discovery rate (FDR) method. In order to minimise false-negative interpretation of time course experiments (without increasing sample sizes post hoc), individual p values were reported with no formal correction for multiple comparisons as recommended elsewhere.25 Predefined samples sizes of approximately 8–12 mice/group were targeted, representing a pragmatic balance between statistical power (based on estimations of variation and effect size observed in pilot experiments), and animal breeding constraints. Statistical tests were performed with GraphPad Prism V.7.0 and Partek Genomics Suite. Specific statistical tests are presented in figure legends. All tests were performed two sided, and a significance level of p≤0.05 was considered statistically significant.
An expanded version of the materials and methods is provided as an online supplementary data.
Supplementary file
Results
Plasma levels of ANGPT2 are significantly elevated in patients with ALI/ARDS and in ANGPT2OVR transgenic mice
We first sought to characterise the plasma levels of ANGPT2 in a cohort of healthy subjects and patients with ALI/ARDS (secondary to septic shock) to define the level and variability associated with relevant clinical plasma specimens (figure 1A). At time of admission to the intensive care unit, patients with ALI/ARDS showed significantly higher plasma levels of ANGPT2 compared with healthy subjects (median (IQR), ALI/ARDS: 17.5 (7.5–35.1) ng/mL vs healthy: 2.4 (1.6–3.1) ng/mL) (figure 1B), which remained elevated at 6 hours, 12 hours, 24 hours, 72 hours and 7 days post admission. Plasma levels of ANGPT2 were quite variable between patients with ALI/ARDS, ranging from 4 to 130 ng/mL. These plasma levels of ANGPT2 are consistent with several previous independent studies of patients with direct or indirect ALI/ARDS,17 26–28 which have reported median levels ranging from 15 to 20 ng/mL and maximal levels up to 115 ng/mL (online supplementary Figure S1). By comparison, transgenic ANGPT2OVR mice exhibited median hANGPT2 plasma levels of 213 ng/mL assessed at baseline and confirmed up to 72 hours after exposure to the bacterial endotoxin, LPS, demonstrating very robust expression and secretion into the systemic circulation (figure 1C). Littermate controlstTA lacking the hANGPT2 responder gene showed no detectable levels of hANGPT2 in plasma (figure 1C). Plasma levels of mouse ANGPT2 were relatively stable following LPS exposure, with only a modest (~2-fold) and transient decline at 3 hours and 6 hours post-LPS (online supplementary figure S2). Thus, systemic hANGPT2 overexpression provides elevated circulating levels of ANGPT2 independent of LPS-induced lung injury. In addition, plasma levels of murine ANGPT2 were slightly higher in ANGPT2OVR versus controltTA mice, though the difference only reached statistical significance at 3 hours and 24 hours post-LPS (online supplementary figure S2).

Circulating levels of human ANGPT2 are elevated in patients with ALI/ARDS and in ANGPT2OVR transgenic mice. (A) Characteristics of healthy subjects and patients with indirect ALI/ARDS (secondary to septic shock). (B) Plasma levels of ANGPT2 assessed in healthy subjects (n=12) and patients with indirect ALI/ARDS (n=8) at several time points following admission to the ICU (n=4–7/time point, subject to sample availability). Plasma specimens were not available for all time points. Kruskal-Wallis and Dunn’s multiple comparison adjusted *p<0.05, **p<0.01, ***p<0.001 versus healthy control group. (C) Plasma levels of hANGPT2 expressed in transgenic mice with and without LPS exposure (n=5–8 mice/group/time point). hANGPT2 was ND in control (tTA) mice lacking the hANGPT2 responder gene. Two-way analysis of variance and Bonferroni multiple comparison adjusted ***p=0.0001, ****p<0.0001 versus ctrltTA group. Box plots denote median, IQR and min/max. ALI, acute lung injury; ANGPT2, angiopoietin-2; ARDS, acute respiratory distress syndrome; FiO2, fractional inspired oxygen; hANGPT2, human ANGPT2; ICU; intensive care unit; LPS, lipopolysaccharide; ND, not detectable; PaO2, partial pressure arterial oxygen.
LPS-induced pulmonary inflammation is exacerbated in ANGPT2OVR mice
ALI was induced via endotracheal delivery of LPS, and an initial dose–response experiment was conducted in controltTA mice to define an LPS dose that would induce inflammation but not saturate the biological readout of total cell infiltration of the alveolar air space. This was an important consideration to reduce the likelihood of potential false-negative outcomes associated with a saturated biologic response to LPS, since we hypothesised that systemic ANGPT2 overexpression would increase pulmonary inflammation. A 1 mg/kg LPS dose induced a significant increase (~6-fold, p<0.05) in total cell levels in BAL 48 hours postdelivery (compared with sham mice) but did not saturate the response as demonstrated by a linear increase in cell infiltration at higher doses (figure 2A). In subsequent time course experiments, total and differential cell counts in BAL were assessed over a range of early and late time points including 3 hour, 6 hour, 24 hours, 48 hours and 72 hours post-LPS exposure. Total cell, neutrophil and macrophage/monocyte cell counts were significantly elevated in the BAL of ANGPT2OVR mice versus controlstTA at 48 hours post-LPS exposure (p<0.05, figure 2B), consistent with a pro-inflammatory role for ANGPT2. Four proinflammatory cytokines were also measured in the BAL fluid at both an early (6 hours) and late (48 hours) time point following LPS-induced injury (figure 2C). The levels of interleukin (IL)-6, IL-1β and monocyte chemoattractant protein-1 (MCP-1) were significantly higher (p<0.05) in ANGPT2OVR mice versus controlstTA specifically at the 6 hours endpoint, again supporting a proinflammatory role for the elevated circulating levels of ANGPT2 in this model. No significant difference in tumour necrosis factor-alpha (TNF-α) levels were observed at 6 hours or 48 hours.

Inflammatory cell infiltration and cytokine levels are elevated in the alveolar air space of LPS-exposed ANGPT2OVR mice. (A) Bronchoalveolar lavage (BAL) total cell counts at different LPS doses in controltTA mice 48 hours after intratracheal LPS delivery (n=3–5 mice/LPS dose). Data represent mean±SEM. Red lines denote linear trend line and error (dotted lines). (B) BAL cell counts at different time points following LPS-induced lung injury. White squares denote controltTA mice, and grey circles denote ANGPT2OVR mice. Time 0 hour (no LPS): n=7 mice/group, 3 hours: n=7–8 mice/group, 6 hours: n=9–10 mice/group, 24 hours: n=9–10 mice/group, 48 hours: n=11–14 mice/group and 72 hours: n=9–10 mice/group). Data represent two independent animal experiments per time point. Red horizontal line and error bars denote mean±SEM. Two-way analysis of variance and Fisher’s LSD test. p values are shown unadjusted for multiple comparisons. (C) Proinflammatory cytokine levels in BAL fluid at two different time points following LPS exposure. Time 6 hours: n=9–12/group, 48 hours: n=11/group. Data represent two independent animal experiments per time point. White squares denote controltTA mice, and grey circles denote ANGPT2OVR mice. Red horizontal line and error bars denote mean±SEM. Unadjusted p values are shown. ANGPT2, angiopoietin-2; IL, interleukin; LPS, lipopolysaccharide; MCP-1, monocyte chemoattractant protein-1; TNF-α, tumour necrosis factor-alpha.
LPS-induced pulmonary vascular leak is not exacerbated in ANGPT2OVR mice
Pulmonary vascular leak was initially assessed by measuring the total protein level in BAL fluid, which is expected to rise with increasing vascular permeability. However, while total protein levels in BAL progressively increased in a time-dependent response to LPS exposure, no significant increase was observed in ANGPT2OVR mice compared with controlstTA (figure 3A). Importantly, the possibility of a false-negative result caused by saturation of the biological response to LPS is unlikely, because the LPS dose was confirmed to induce only submaximal vascular leak in LPS-titration experiments and was near the lower end of the linear dynamic range of BAL total protein measurements (figure 3B). Pulmonary oedema, as evidenced by the lung wet/dry weight ratio, was significantly increased at 24 hours and 48 hours post-LPS exposure (p<0.05 vs mice that did not receive LPS); however, there were no significant differences between the ANGPT2OVR mice and controlstTA at either time points (figure 3C). BAL levels of the two plasma proteins IgM and albumin, which represent more specific and sensitive markers of vascular permeability, were also measured to further assess pulmonary leak (figure 3D). Interestingly, while BAL levels of IgM and albumin increased time-dependently in response to LPS exposure, consistent with increased vascular permeability in the LPS model, the levels of these specific proteins showed marked attenuation in ANGPT2OVR mice compared with controlstTA at the 24 hours and 48 hours endpoints (p<0.05). These results suggest that high plasma levels of ANGPT2 might provide partial protection against LPS-induced vascular leak, which would support a previous report in which ANGPT2 was also shown to decrease vascular leak in mice exposed to another type of inflammatory agent.15

LPS-induced pulmonary vascular leak is not exacerbated in ANGPT2OVR mice. (A) Bronchoalveolar lavage (BAL) levels of total protein at different time points following LPS-induced lung injury. (B) Pilot experiment demonstrating subsaturating total protein levels in BAL of controltTA mice 48 hours after LPS exposure (n=3–5 mice/LPS dose). Error bars denote SEM. Red line shows linear trend line and associated error (dotted lines). (C) Assessment of pulmonary oedema by the lung wet/dry weight ratio. One-way analysis of variance (ANOVA) and Dunnet’s multiple comparison test. *p<0.05, **p<0.01, ****p<0.0001 versus no LPS group. (D) BAL levels of IgM and albumin at different time points following LPS exposure. White squares denote controltTA mice, and grey circles denote ANGPT2OVR mice. Two-way ANOVA and Fisher’s LSD test (p values unadjusted for multiple comparisons). For panels (A), (C) and (D), red horizontal lines and error bars denote mean±SEM. Sample sizes in time course experiments were time 0 hour (no LPS): n=7 mice/group, 3 hours: n=7–8 mice/group, 6 hours: n=9–10 mice/group, 24 hours: n=9–10 mice/group, 48 hours: n=11–14 mice/group, 72 hours: n=9–10 mice/group). Data represent two independent animal experiments per time point. ANGPT2, angiopoietin-2; LPS, lipopolysaccharide.
Systemic overexpression of ANGPT2 neither promotes nor protects against LPS-induced mortality
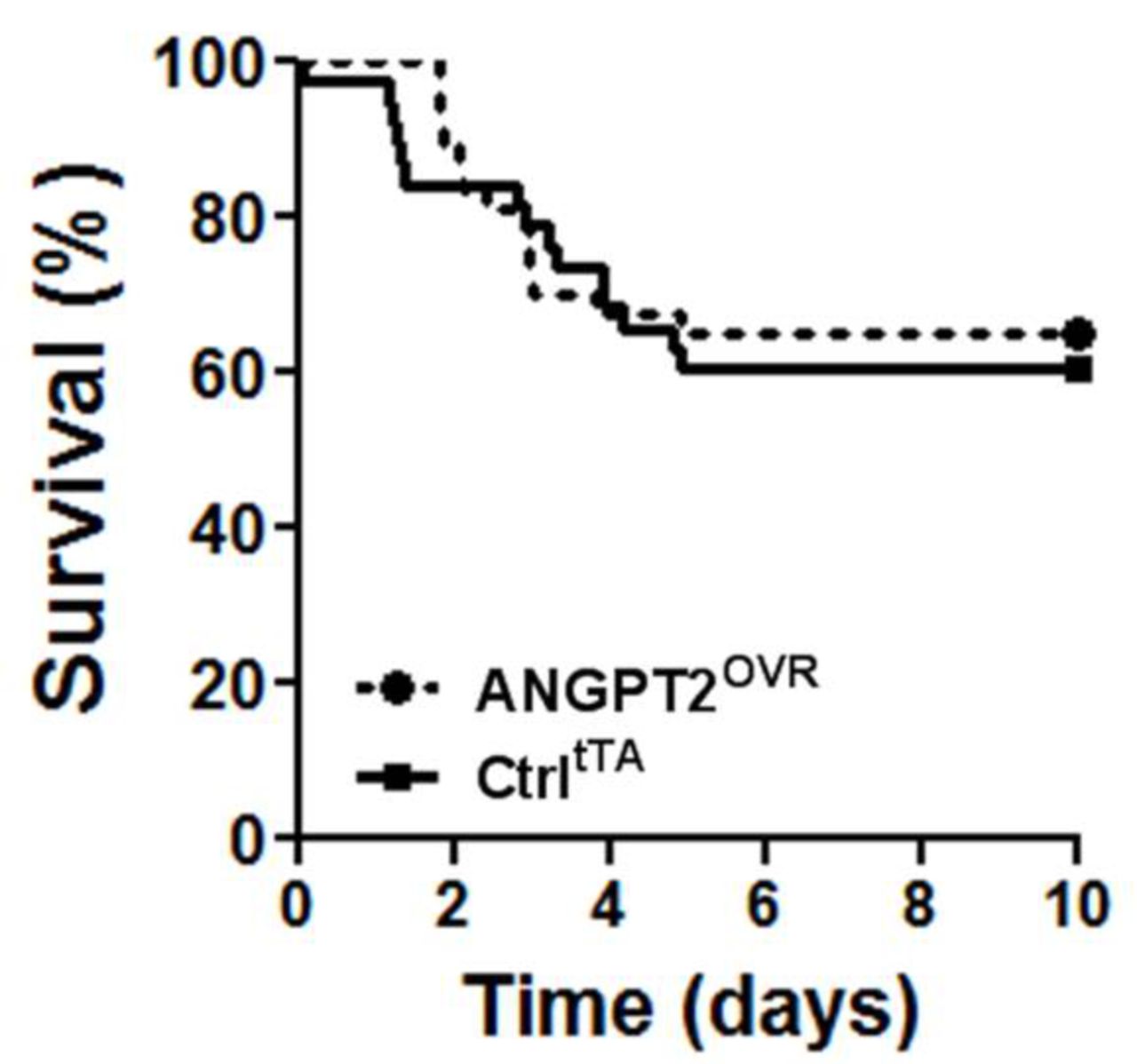
In light of the contrasting effects on lung vascular inflammation and leak, we sought to determine whether the net effect of high circulating ANGPT2 levels would be detrimental or beneficial to the survival of mice with severe LPS-induced lung injury (figure 4). However, no significant difference in survival was observed in this model: 39% of controltTA mice (14 of 37 mice) and 37% (14 of 38 mice) of ANGPT2OVR mice exhibited terminal morbidity by day 5, with no further events observed up to the predefined experimental endpoint on day 10.
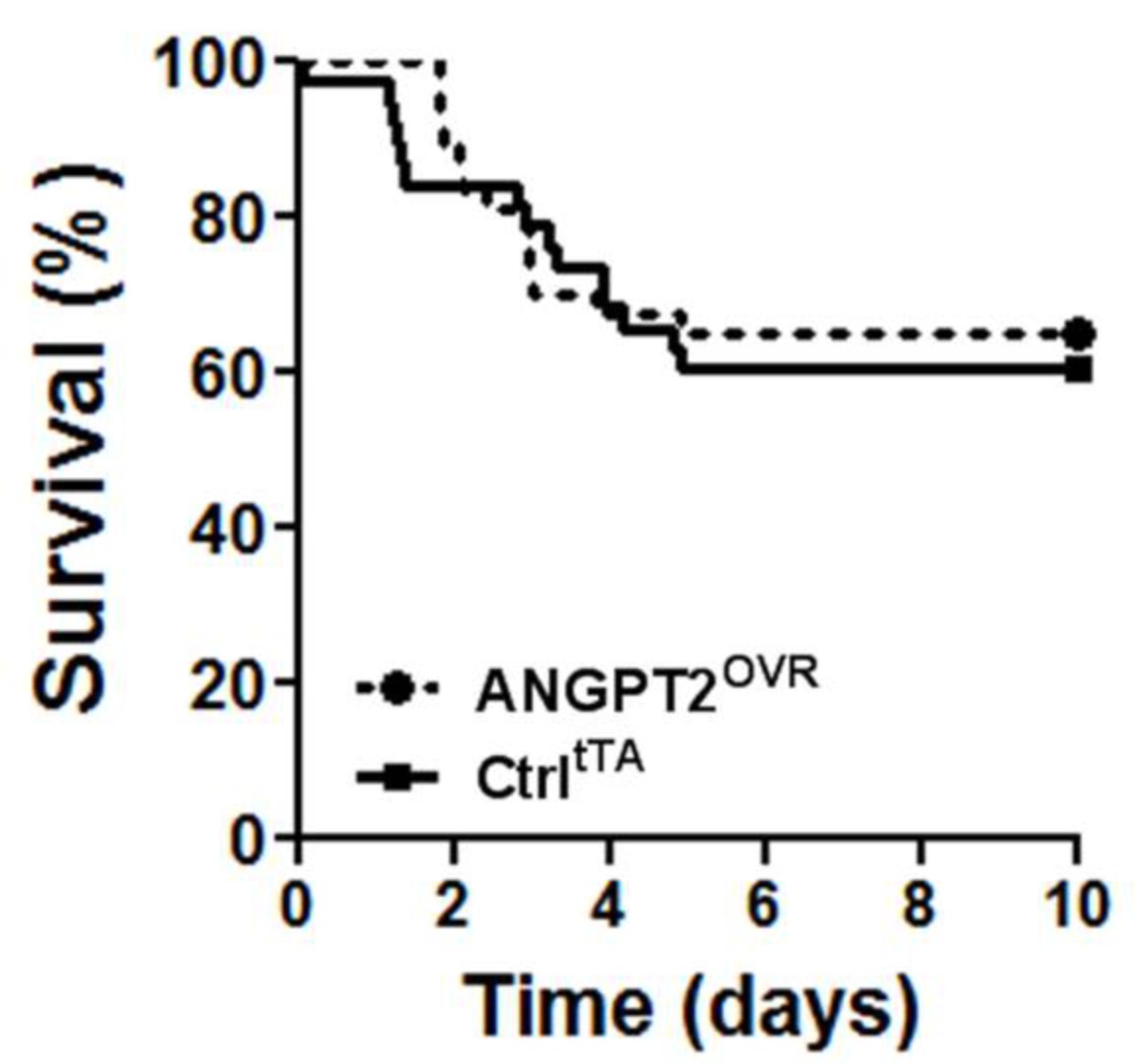
Survival following high-dose LPS challenge. Intratracheal administration of 10 mg/kg LPS in ANGPT2OVR and controltTA (Ctrl) littermate mice. Survival curves were not significantly different by log-rank test (n=37–38 mice/group, from five independent animal experiments). ANGPT2, angiopoietin-2; LPS, lipopolysaccharide.
Pulmonary changes in angiopoietin expression and TIE2 receptor activation
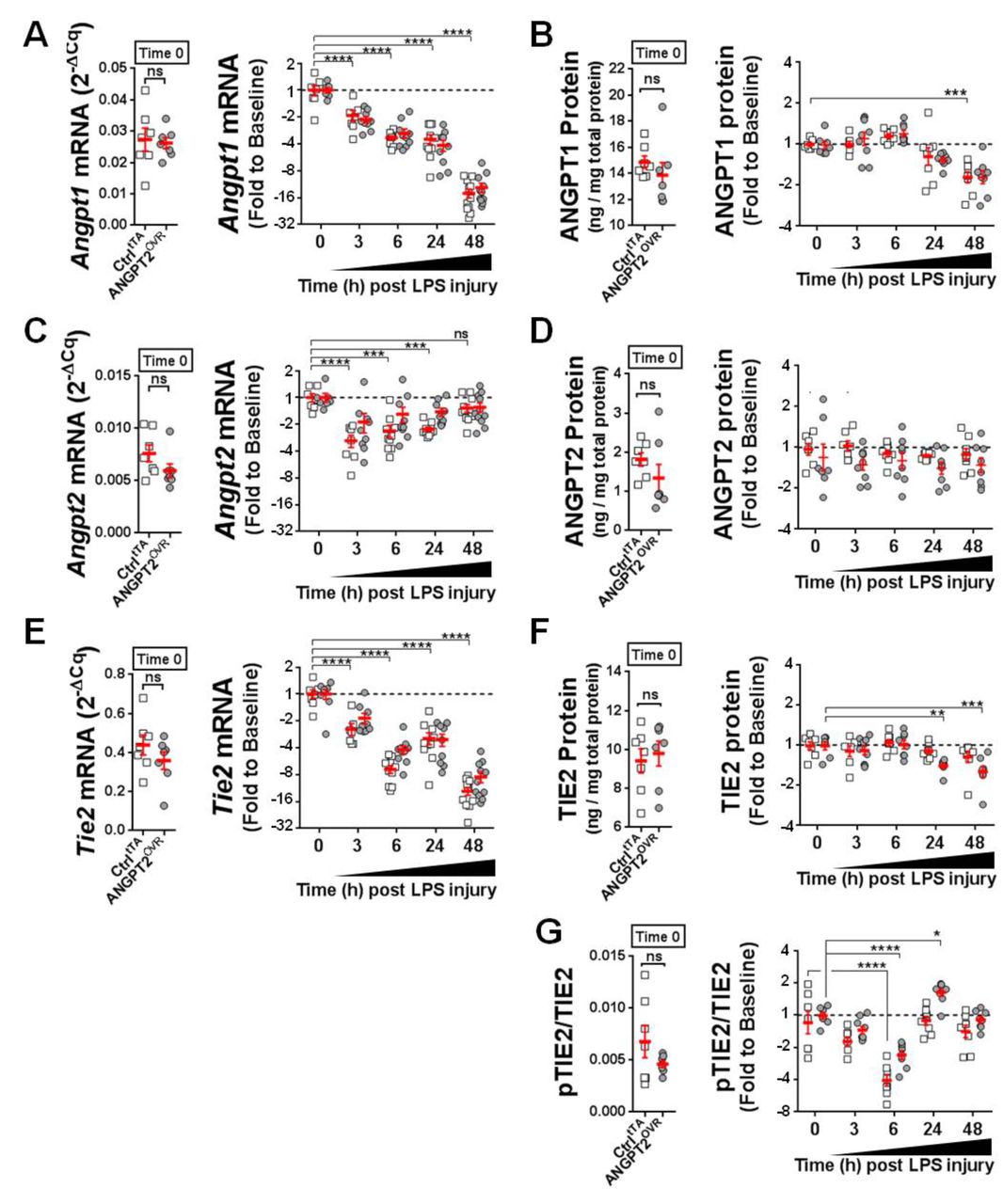
The pulmonary regulation of endogenous mouse Angpt1, Angpt2 and Tie2 expression was examined to gain further insight into the potential context-dependent actions of Angpt2. 13 14 At baseline (ie, prior to LPS exposure), mouse Angpt1 mRNA levels were ~4-fold higher than Angpt2 (figure 5A vs figure 5C); however, Angpt1 expression progressively decreased (up to ~16-fold by 48 hours post-LPS exposure; figure 5A), while the expression of Angpt2 remained relatively fixed near baseline levels, with only a transient decrease (~2–4 fold, p<0.05) at earlier time points (figure 5C). A similar pattern in angiopoietin expression was also observed at the protein level, though the rate and magnitude of change were more modest (figure 5B,D). Lung mRNA levels of Tie2 showed a progressive and significant decline after LPS exposure (p<0.0001, figure 5E), which was also detectable at the protein level, although to a lesser extent and with delayed kinetics (figure 5F). Of note, no significant differences were detected between controltTA and ANGPT2OVR mice in ANGPT1, ANGPT2 and TIE2 levels at the mRNA or protein levels at baseline and after LPS exposure (figure 5). Pulmonary transcript levels of Vegfa were also compared between ctrltTA and ANGPT2OVR mice (online supplementary figure S3), given its importance in vascular permeability29 30 and potential for crosstalk with TIE2 signalling pathways31; however, no significant differences were observed between transgenic mouse groups at baseline or after LPS exposure. We next sought to investigate pulmonary TIE2 receptor activation using the ratio of pTIE2 to total TIE2 as a readout (figure 5G). An initial decline in pTIE2/TIE2 was observed from baseline to 6 hours post-LPS exposure (−2.3 mean fold change for ANGPT2OVR and −3.9 fold change for ctrltTA, p<0.0001 vs baseline), followed by a recovery to baseline levels by 24 hours in both ctrltTA and ANGPT2OVR mice. Importantly, however, in ANGPT2OVR but not controltTA mice, TIE2 phosphorylation was enhanced 1.7-fold above baseline levels at 24 hours (p<0.05), consistent with a potential time-dependent agonist role for circulating ANGPT2.

Changes in pulmonary levels of ANGPT1, ANGPT2, TIE2 and pTIE2/TIE2. (A, C andE) Lung mRNA levels of mouse Angpt1, Angpt2 and Tie2 in controltTA mice (open squares) and ANGPT2OVR mice (grey circles). Left panel shows mRNA levels measured by RT-qPCR at baseline (ie, time 0, no LPS) presented in relative expression units (normalised to the geomean of Gapdh and β-actin). Right panel shows the change in mRNA levels after LPS exposure. Data are presented as fold change to baseline and log2 transformed; n=7–11 mice/genotype group/time point). (B, D and F) Lung protein levels of mouse ANGPT1, ANGPT2 and TIE2 measured by ELISA. Left panel shows protein levels at baseline normalised to total protein levels. Right panel shows the change in protein levels after LPS exposure. Data are presented as fold change to baseline and log2 transformed; n=7–9 mice/genotype group/time point. (G) Ratio of pTIE2 to total TIE2 protein at baseline (left panel) and after LPS exposure (right panel, presented as fold change to baseline and log2 transformed) n=7–9 mice/genotype group/time point. Error bars denote mean±SEM. For clarity, statistical tests for LPS-induced changes in mRNA and protein levels are only shown for the controltTA group (two-way analysis of variance (ANOVA) and Dunnett’ s multiple comparison test; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 vs time 0), with the exception of panel F where significant differences were only detected in the ANGPT2OVR group. No significant differences between genotype groups were detected by two-way ANOVA and Sidak’s multiple comparison test. ANGPT2, angiopoietin-2; LPS, lipopolysaccharide; pTIE2, phospho-TIE2.
Lung transcriptional profiling reveals dynamic temporal regulation of diverse vascular genes in response to LPS-induced injury
To gain insight into underlying molecular mechanisms, we measured the pulmonary mRNA expression of 84 genes known to be involved in critical signalling pathways related to vascular biology including angiogenesis, vascular tone, inflammation, apoptosis, cell adhesion, coagulation and platelet activation. Because the actions of ANGPT2 are known to be highly context dependent, we first sought to establish a framework to understand the temporal regulation of these genes in response to LPS, independent of high circulating ANGPT2 levels. Therefore, transcript levels were first investigated in controltTA mice at baseline (no LPS) and two representative time points after LPS exposure to assess both an early (6 hours) and later (48 hours) phase of lung injury. All but one gene showed a significant change in expression following LPS exposure (FDR <0.05, ranging from 43-fold down to 190-fold up versus baseline (no LPS) group; figure 6A and Table E1 in the online supplementary data), and their temporal regulation was evident by the distinct expression patterns observed at 6 hours and 48 hours, as reflected in the hierarchical clustering of mice (figure 6A). Proinflammatory cytokines/chemokines such as IL-1b, Ccl2, TNF-α, Cxcl1 and Cxcl2 were the most highly upregulated genes (from 23-fold to 190-fold elevated), and several key genes linked to vascular integrity, such as Cdh5, Ocln and Angpt1, were all significantly downregulated (from −7 fold to −44 fold decreased), consistent with the lung inflammation and vascular leak observed in this LPS model. The lung transcript profiles also revealed differences in how gene expression within different functional groups of related genes were regulated time-dependently between the early and late phases of lung injury. The inflammation group showed the highest proportion of genes with peak changes in expression at 6 hours, and the most balanced distribution, with 46% and 54% of genes showing maximal changes at 6 hours and 48 hours, respectively (figure 6B). By comparison, peak changes in the expression of cell adhesion-linked genes (that may also play important roles in barrier function) was skewed markedly towards the later phase of lung injury, with 35% and 65% of these genes peaking at 6 hours and 48 hours, respectively. Genes associated with apoptosis, platelet activation and vascular tone were the most disproportionately regulated, with 79%–88% showing peak changes in expression at 48 hours.
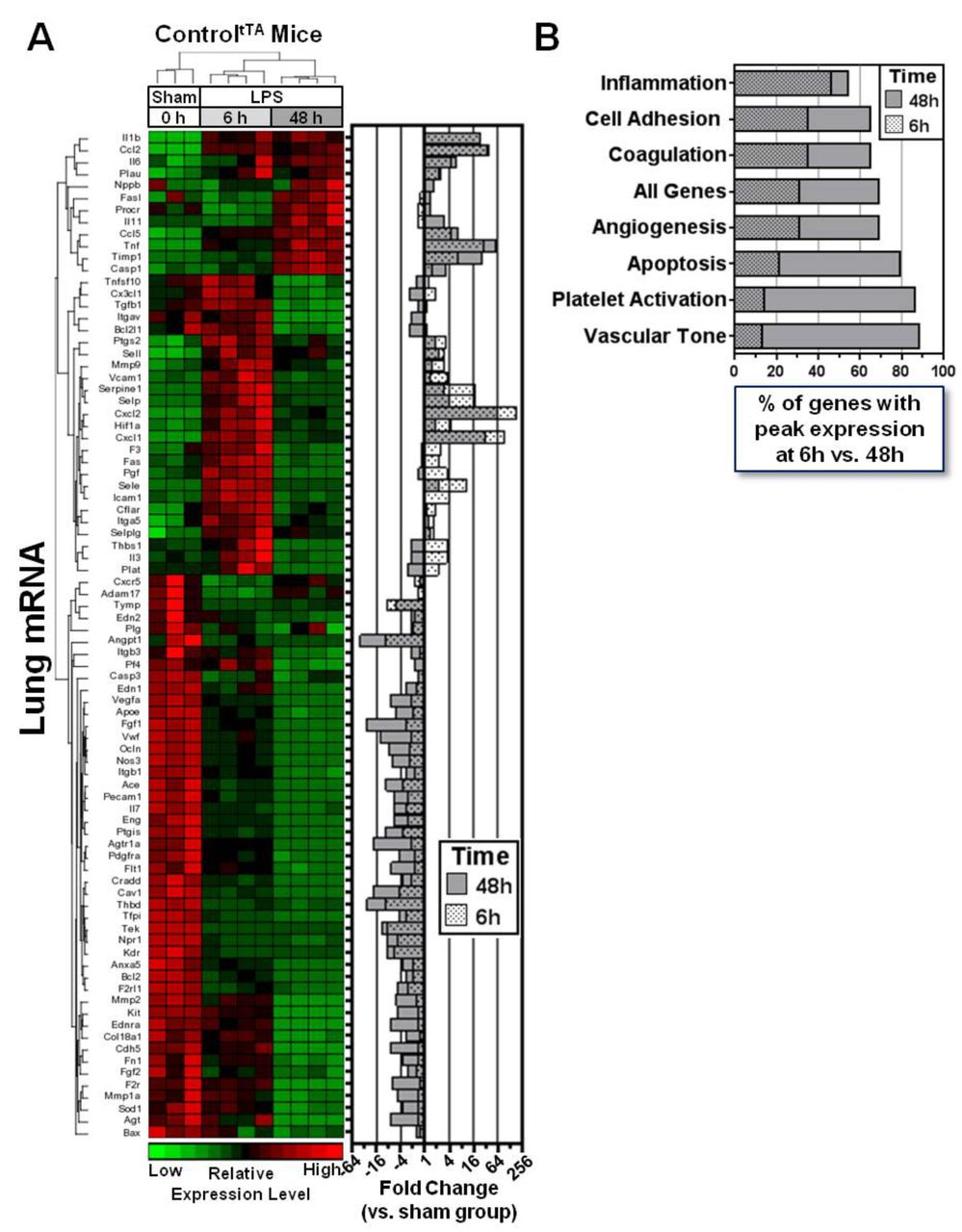
Lung transcriptional profiling reveals dynamic temporal regulation of vascular genes in LPS-injured mice. (A) Hierarchical clustering of gene expression patterns in sham and LPS-exposed controltTA mice at 6 hours and 48 hours. The heatmap shows relative expression levels of 84 predefined genes that were assessed by PCR array. The right panel shows the mean fold change in expression of each gene at both 6 hours and 48 hours post-LPS versus the sham group. Data for 6 hours and 48 hours are superimposed on the graph to facilitate viewing. (B) The graph shows how gene expression across different functional groups is regulated between the early and later phases of lung injury, based on the percentage of genes that showed peak changes in expression (up or down) at 6 hours or 48 hours post-LPS exposure. LPS, lipopolysaccharide.
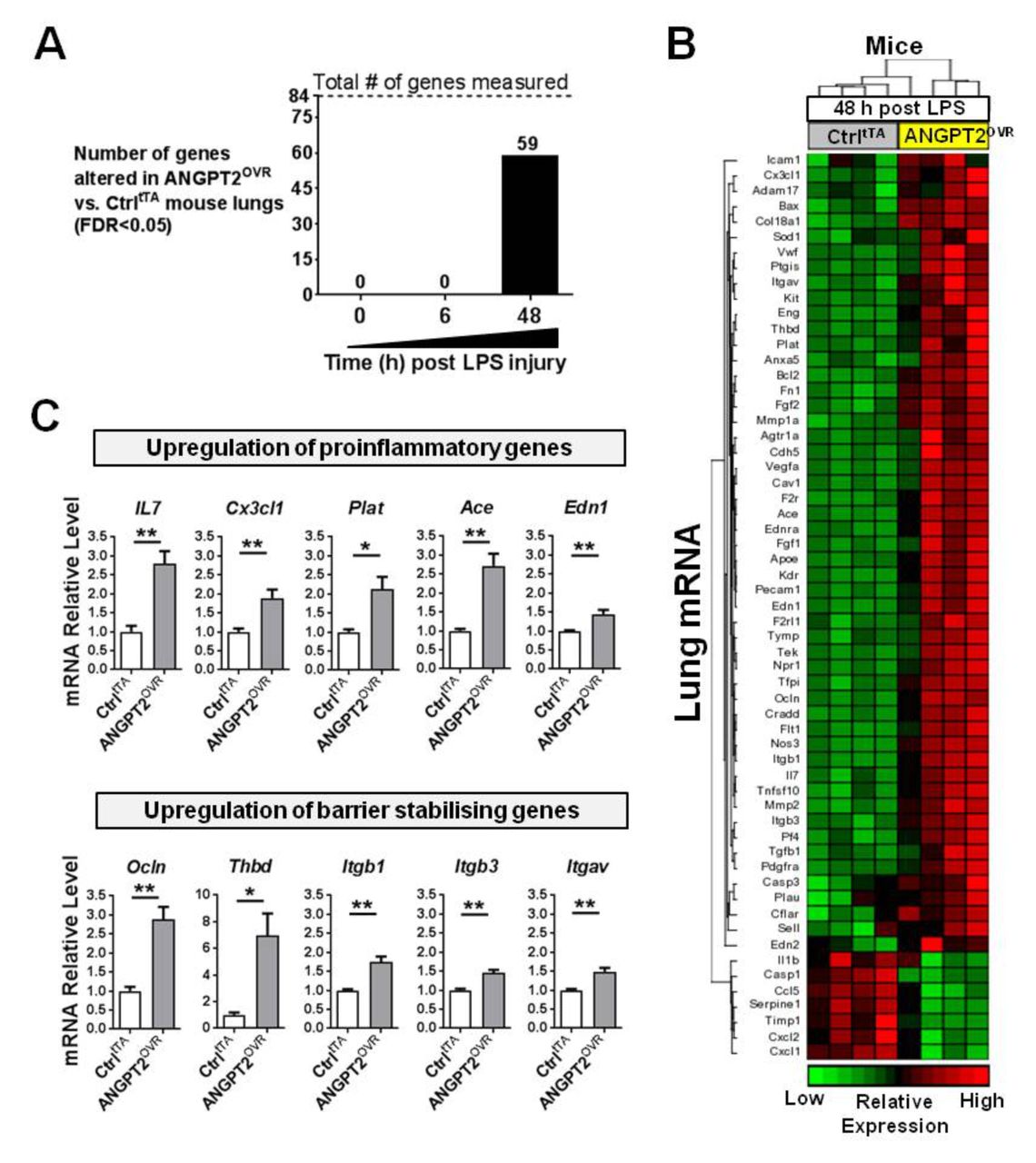
Proinflammatory and barrier-stabilising genes are time-dependently upregulated in ANGPT2OVR mice after LPS injury
We next sought to evaluate the effects of elevated plasma ANGPT2 levels on pulmonary gene expression before and after LPS exposure. Under baseline physiological conditions (ie, no LPS exposure), there were no significant differences in the lung transcript profile between ANGPT2OVR and controltTA mice (table E2 in the online supplementary data), suggesting that even high plasma levels of ANGPT2 may not alter lung vascular biology under normal physiological conditions. Similarly, at 6 hours post-LPS exposure, no significant differences in lung gene expression were detectable between ANGPT2OVR and controltTA mice (table E3 in the online supplementary data). However, at 48 hours post-LPS exposure, ANGPT2OVR mice exhibited significant alterations in the expression of 59 genes (FDR <0.05 vs controltTA mice; figure 7A,B and table E4 in the online supplementary data), including representatives from each of the functional groups described previously. The magnitude of change in transcript levels triggered by systemic ANGPT2 overexpression was modest (ie, mean change; −1.6-fold down and 2.4-fold up) compared with the changes induced by LPS exposure (ie, mean change; −7.4 fold down and 16.6-fold up at 48 hours), which is consistent with the moderate effects ANGPT2OVR mice showed on neutrophil infiltration (figure 2) and vascular leak in the lungs (figure 3). Interestingly, the proinflammatory mediators that were increased by LPS exposure in controltTA mice (ie, lL-1b, Ccl2, Tnfα, Cxcl1 and Cxcl2) were not increased further in the ANGPT2OVR mice at 48 hours; however, a separate set of genes known to promote neutrophil adhesion and recruitment such as IL-7,32 endothelin-1 (Edn1),33 plasminogen activator tissue (Plat),34 ACE35 and chemokine (C-X3-C motif) ligand 1 (also known as fractalkine, Cx3cl1) were significantly elevated (figure 7C). This suggests that multiple molecular mechanisms may act to potentiate pulmonary inflammation. ANGPT2OVR mice also showed increased expression of several genes that were decreased by LPS exposure and that are known to promote endothelial barrier integrity, such as occludin (Ocln), thrombomodulin (Thbd) and integrins (Itgb1, Itgb3 and Itgav) (figure 7C).

ANGPT2OVR mice show time-dependent alterations in pulmonary gene expression at 48 hours post-LPS exposure. (A) Number of genes significantly altered by systemic ANGPT2 overexpression before (time 0) and after LPS exposure. Eighty-four predefined genes linked to endothelial cell biology were measured by RT-qPCR in lung tissue. (B) Heatmap showing relative expression levels of 59 genes differentially expressed between controltTA (Ctrl) and ANGPT2OVR mice (FDR<0.05). (C) Bar graphs show relative mRNA levels of several genes that were significantly upregulated in ANGPT2OVR mice 48 hours post-LPS, with functional roles in neutrophil adhesion and recruitment (ie, proinflammatory) or endothelial barrier stabilisation. *p<0.05, **p<0.01 versus CtrltTA. Data are presented as mean±SEM (n=4/group). ANGPT2, angiopoietin-2; LPS, lipopolysaccharide.
In vivo neutralisation of circulating ANGPT2 partially reverses the effects on lung vascular inflammation and leak in LPS-injured mice
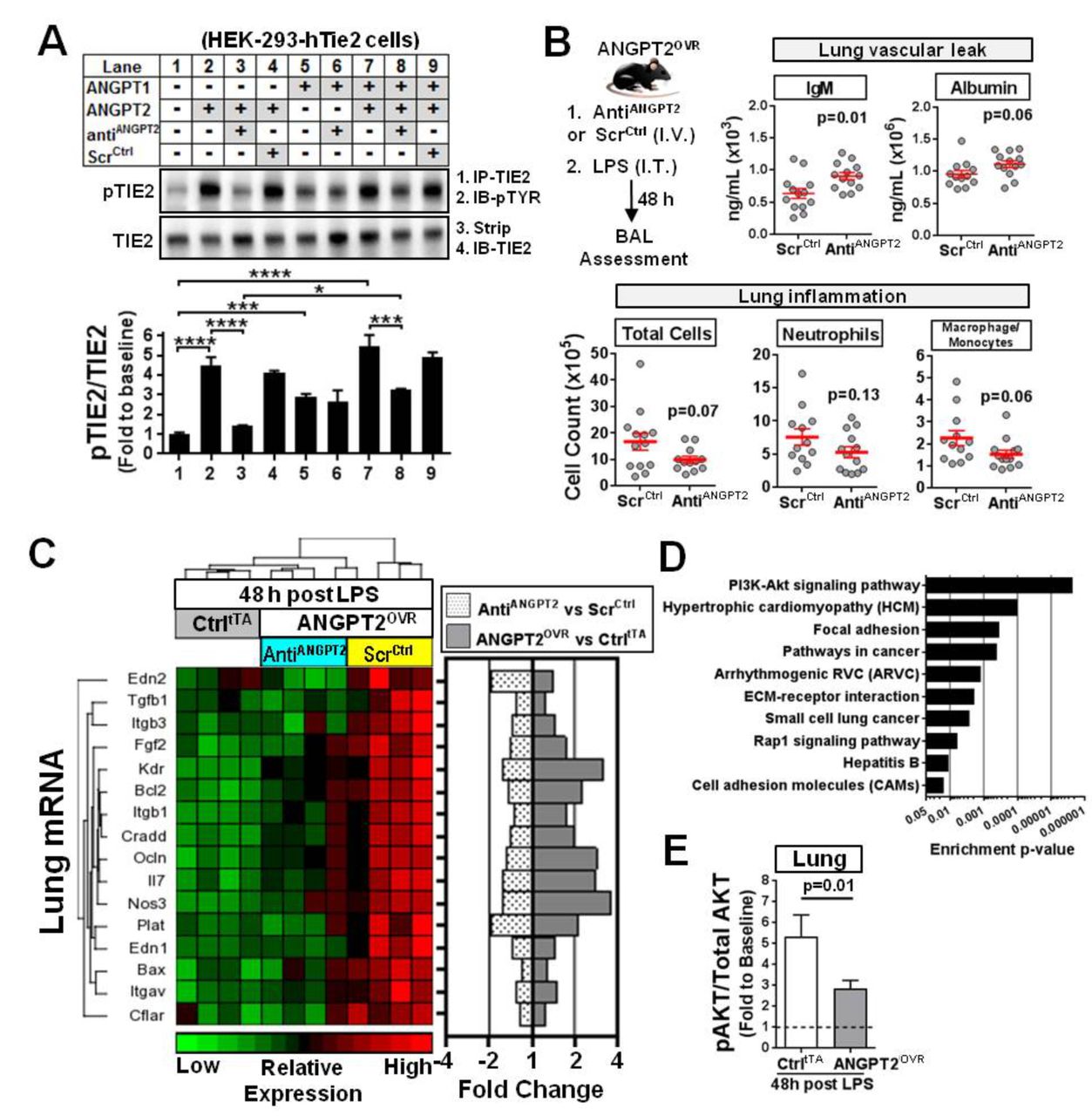
To confirm whether the high circulating levels of ANGPT2 directly contributed to the observed effects on lung vascular inflammation and leak, an aptamer-based inhibitor24 was used to neutralise the extracellular circulating ANGPT2 in ANGPT2OVR mice exposed to LPS. This aptamer has previously been shown to bind to hANGPT2 protein with high affinity and specificity.24 The ability of the aptamer to selectively block ANGPT2-mediated TIE2 activation, but not ANGPT1-mediated TIE2 activation, was first confirmed in vitro using a TIE2 receptor phosphorylation assay (figure 8A). In a cultured EC system, ANGPT2 does not induce robust phosphorylation of TIE2 except at very high concentrations24; therefore, a more practical assay system was adopted to validate the inhibitory action of the aptamer using HEK-293 cells that ectopically express TIE2, as described previously.24 The antiANGPT2 aptamer effectively blocked TIE2 phosphorylation induced by recombinant human (rh) ANGPT2 (p<0.0001) but did not block TIE2 phosphorylation induced by rhANGPT1 (figure 8A, lanes 1–6). This effect was specific to the aptamer, as no inhibition of TIE2 phosphorylation was observed with a scrambled-sequence control oligonucleotide. To further delineate the potential mechanism by which the aptamer blocks TIE2 phosphorylation, we also examined its neutralising effects in the presence of both rhANGPT2 and rhANGPT1 simultaneously (figure 8A, lanes 7–9). This is important because the aptamer could prevent ANGPT2 from binding to the TIE2 receptor, or simply prevent dimerisation of the receptor (which is necessary for TIE2 phosphorylation) after ANGPT2 binds to TIE2; however, the former mechanism would be more effective since it would not interfere with interactions between ANGPT1 and TIE2. In the presence of both rhANGPT2 and rhANGPT1, the level of TIE2 phosphorylation was significantly attenuated in cells treated with the aptamer (p<0.001 vs no aptamer, figure 8A, lane 8 vs 7), but still significantly higher (p<0.05) than cells treated with only rhANGPT2 and aptamer (figure 8A, lane 3), and on par with cells treated only with ANGPT1 and aptamer (figure 8A, lane 6). Collectively, these results support a mechanism in which the aptamer selectively blocks ANGPT2-mediated TIE2 activation and does not interfere with the interaction between ANGPT1 and TIE2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vitro and in vivo neutralisation of ANGPT2. (A) In vitro Tie2 phosphorylation assay demonstrates selective blockade of ANGPT2-mediated TIE2 receptor activation by an anti-hANGPT2 aptamer. HEK293 cells ectopically expressing human TIE2 were treated with various combinations of recombinant human ANGPT1 (rhANGPT1) (3 nM), rhANGPT2 (15 nM), the anti-ANGPT2 aptamer (AntiANGPT2; 15 nM) or scrambled-sequence control RNA molecule (ScrCtrl, 15 nM). PhosphoTIE2 (pTIE2) was measured by immunoprecipitation (IP) for TIE2 followed by immunoblotting (IB) for phosphotyrosine (pTYR). Blots were stripped and reprobed for TIE2. Representative blots are shown. Summary data are presented as mean±SEM and expressed relative to baseline (lane 1) (n=3–5 independent experiments). One-way analysis of variance and Tukey’s multiple comparison adjusted *p<0.05, ***p<0.001, ****p<0.0001. (B) In vivo effects of anti-ANGPT2 aptamer treatment in ANGPT2OVR mice following LPS-induced lung injury. ANGPT2OVR mice received a intravenous (IV) tail injection of aptamer (AntiANGPT2) or scrambled control (ScrCtrl) just prior to intratracheal (IT) LPS challenge. BAL levels of different cell and plasma proteins were measured 48 hours later. Data are presented as mean±SEM (n=12–13 mice/group, from three independent animal experiments). Exact p values are shown for unpaired two-sided t-tests. (C) Hierarchical clustering and heatmap showing relative expression and fold change levels of 16 genes that were significantly increased in ANGPT2OVR (ScrCtrl) mice versus CtrltTA mice 48 hours post-LPS challenge and attenuated by AntiANGPT2 aptamer treatment (FDR <0.05). The mean fold change in expression of each gene is shown on the right for different group comparisons. (D) Top 10 pathways significantly enriched in genes altered by circulating ANGPT2 as determined by gene set enrichment analysis. (E) The ratio of phosphoAKT (Ser473) to total AKT protein in the lungs. Data are presented as mean±SEM (n=10–13 mice/group, from two independent experiments). Exact p value shown for Mann-Whitney U test. ANGPT2, angiopoietin-2; AKT, protein kinase B; ECM, extracellular matrix; FDR, false discovery rate; hANGPT2, human ANGPT2; LPS, lipopolysaccharide.
The in vivo effects of neutralising circulating ANGPT2 in ANGPT2OVR mice was then evaluated by intravenous administration of the aptamer or scrambled-sequence control just prior to LPS exposure. A non-significant but consistent trend towards decreased BAL levels of total cells (1.7-fold decrease, p=0.07), neutrophils (1.4-fold decrease, p=0.13) and macrophage/monocytes (1.5-fold decrease, p=0.06) was observed in the ANGPT2OVR mice treated with the antiANGPT2 aptamer (compared with ANGPT2OVR mice treated with the scrambled-sequence control; figure 8B). However, indices of vascular leak were concordantly increased in LPS-exposed ANGPT2OVR mice that received the antiANGPT2 aptamer as reflected in the increased BAL IgM (p=0.01) and albumin (p=0.06) levels (figure 8B), again consistent with a possible vasculoprotective role for circulating ANGPT2 in this model.
Targeted lung transcriptional profiling of 84 candidate genes was conducted once again to derive further mechanistic insight. A total of 16 genes were both significantly increased in LPS-exposed ANGPT2OVR mice (vs LPS-exposed controltTA mice) and subsequently attenuated by the antiANGPT2 aptamer treatment (FDR <0.05, figure 8C). Of note, aptamer-mediated neutralisation of circulating ANGPT2 in LPS-exposed ANGPT2OVR mice significantly blocked (FDR <0.05) the upregulation of genes linked to neutrophil adhesion and recruitment (ie, IL-7, Edn1 and Plat), as well as endothelial barrier integrity (ie, Ocln, Thbd, Itgav, Itgb1 and Itgb3). In addition, aptamer treatment also attenuated the expression of several apoptosis-linked genes that could potentially exacerbate the inflammatory response through cell death mechanisms. To gain additional insight into the potential signalling pathways affected by the altered genes, we performed a gene set enrichment analysis using Partek Pathway. Among the top 10 pathways that were significantly enriched in genes altered by high plasma ANGPT2 levels after LPS injury (figure 8D), several were linked to vascular inflammation and permeability including cell adhesion molecules (rank 10), Rap1 signalling (rank 8), ECM-receptor interaction (rank 6), focal adhesion (rank 3) and PI3K-Akt signalling (rank 1). The highest ranked pathway involved protein kinase B (AKT), which is known to be a central transducer of multiple cell signalling pathways. We therefore examined AKT protein phosphorylation (ie, pAKT/AKT ratio) in the lungs at 48 hours post-LPS. AKT phosphorylation was significantly increased (~5-fold above baseline) in controltTA mice 48 hours post-LPS injury (p<0.05, figure 8E), but this was significantly attenuated in ANGPT2OVR mice (figure 8E), suggesting that ANGPT2 may contribute to the resolution of lung injury through an AKT-dependent mechanism.
Discussion
In this study, we investigated whether elevated plasma levels of ANGPT2 play a role in potentiating the lung vascular injury and mortality associated with ALI/ARDS in patients. Our results show that in transgenic mice with LPS-induced ALI, high plasma levels of ANGPT2 (induced independent of LPS-exposure by hepatocyte-specific overexpression and secretion) exert only modest effects on lung microvascular function, exacerbating inflammation but not vascular leak, with no effect on LPS-induced mortality.
Our finding that elevated plasma levels of ANGPT2 potentiated pulmonary inflammation during LPS-induced lung injury was supported by increases in proinflammatory cells and cytokines in the alveolar air space of mice. However, contrary to our original hypothesis, LPS-induced pulmonary vascular leak and oedema showed no change in ANGPT2OVR mice, based on BAL total protein levels and lung wet/dry weight ratios, respectively. Interestingly, more specific markers of vascular permeability such as IgM and albumin both appeared to be decreased in the BAL of ANGPT2OVR mice compared with controlstTA, suggesting that elevated plasma levels of ANGPT2 might provide some partial protective effects under specific contexts in vivo. At the molecular level, these biological effects were supported by underlying changes in the pulmonary expression of multiple genes, including upregulation of genes known to be important for inflammatory mechanisms involving neutrophil adhesion and recruitment and upregulation of genes involved in endothelial barrier stability via cell–cell and cell–matrix junctional integrity. Interestingly, significant differences in the pulmonary gene expression profiles between ANGPT2OVR and controltTA mice were only evident under pathological (ie, post-LPS exposure) but not physiological conditions, suggesting that high plasma ANGPT2 levels are necessary but not sufficient alone to induce the observed effects on vascular inflammation and permeability, reinforcing the context-dependent pleiotropic actions of ANGPT2. The temporal regulation of these transcriptional changes, which occurred later in the time course of lung injury (at 48 hours post-LPS), coincided with a significant decline in the pulmonary levels of ANGPT1 and TIE2, suggesting a potential coordinated response to vascular injury. These changes have two potential interpretations with important implications. First, previous cell culture studies have shown that ANGPT2 can function as a dose-dependent antagonist of TIE2 when ANGPT1 is present, but as an agonist when ANGPT1 is absent,14 suggesting that ANGPT2 activity could switch between antagonist and agonist as lung injury progresses over time. Thus, we speculate that stable or increased levels of ANGPT2 with context-induced agonist activity could serve as a potential negative feedback mechanism to compensate for decreased endothelial TIE2 signalling caused by the reduction in ANGPT1 and TIE2 levels. Second, the decrease in TIE2 levels could also favour indirect effects mediated through TIE2-independent or endothelial-independent mechanisms, which might contribute to the diverse transcriptional changes observed in the lung. These distinct mechanisms could include ANGPT2 signalling through alternate endothelial receptors such as integrins,36 or alternate TIE2-expressing cells such as neutrophils,37 monocytes38 39 and lung epithelials.40
The magnitude of effects induced by systemic ANGPT2 overexpression was relatively moderate compared with the effects of LPS exposure. Furthermore, broad transcriptional reprogramming was observed in the lungs of ANGPT2OVR mice compared with controlstTA after LPS exposure, which was associated with small changes in many different effector genes related to vessel function and homeostasis. Collectively, this suggests that circulating ANGPT2 may be better suited for fine-tuning the vascular response to lung injury, rather than driving major pathophysiological changes. This role would be consistent with the minimal impact that was observed on LPS-induced mortality in this model and the dynamic effects on pulmonary inflammation and leak. Gene set enrichment analysis identified the multifunctional protein kinase, Akt, which has an established role in blood vessel homeostasis,41 as one of the key cellular transducers of these transcriptional changes. Previous studies have demonstrated that AKT activation may contribute to vascular leak in vivo42 and the development of LPS-induced ALI.43 In the current study, the elevation in lung AKT phosphorylation induced by LPS exposure was significantly attenuated in ANGPT2OVR mice compared with controlstTA at 48 hours, which suggests a potential homeostatic role for ANGPT2.
In the present study, ANGPT2OVR mice showed an enhanced inflammatory response to LPS exposure, which is consistent with the proinflammatory role of ANGPT2 that has been reported previously.44 In contrast to some previous studies that have reported increased vascular leak when rhANGPT2 was injected in healthy mice,45 our results suggest that ANGPT2 may attenuate vascular leak in mice with lung injury. The different physiological settings of these experiments is an important distinction given the increasing evidence that ANGPT2 activity may fluctuate based on the activation state of the endothelium.46 In the context of LPS-induced lung injury, we observed a potential stress-dependent protective effect of ANGPT2. This result is consistent with findings by Yancoupolous’ group, which have previously reported that ANGPT2 can function as an autocrine protective factor in ECs exposed to shear stress and can also inhibit vascular leak induced by inflammatory agents in mice.15 The potential vascular protective effect of ANGPT2 is also supported by a report by Kurniati et al,47 in which ANGPT2 deficient mice were shown to have higher albuminuria than wild-type controls in an intraperitoneal LPS model of acute kidney injury.47 Moreover, these ANGPT2-deficient mice showed reduced endothelial adhesion molecule expression and neutrophil recruitment in the kidney, consistent with a proinflammatory effect of ANGPT2. Thus, the dissociation between inflammation and vascular integrity observed in our study is supported by several independent studies and under various contexts. In a study by Hegeman et al 48, administration of rhANGPT1 in mice with ventilator-induced lung injury decreased inflammation but not vascular leak. Changes in vascular permeability independent of inflammation have also been reported. David et al 49 showed that the synthetic TIE2 agonist, vasculotide, prevented endotoxaemia-induced pulmonary vascular leak in mice but failed to modulate markers of inflammation. In addition, the study by David et al demonstrated that vascular leak was attenuated through a mechanism involving TIE2 activation and signalling. Taken together, these studies suggest that inflammation and vascular leak can be modulated independently, and this dissociation likely involves more than one distinct mechanism. In our study, we speculate that the differential effects of systemic ANGPT2 overexpression could potentially be reconciled through distinct cell type-specific activities. One possibility is that enhanced inflammation in LPS-injured ANGPT2OVR mice could be due to the chemoattractant properties of ANGPT2, which has previously been shown to recruit TIE2-positive monocytes/macrophages and neutrophils,37 38 while decreased vascular leak could be mediated separately through the ANGPT2-dependent activation of endothelial TIE2. Consistent with this idea, Han and colleagues have recently shown that administration of the ANG2-binding and TIE2-activating antibody (ABTAA), which triggers ANGPT2 clustering and subsequent activation of the TIE2 receptor, effectively attenuated LPS-induced pulmonary vascular leak in a sepsis mouse model.50 This is also consistent with the dynamic changes in TIE2 phosphorylation detected in pulmonary tissue in our model, which showed an initial decline in TIE2 activation (ie, pTIE2/TIE2 ratio) from 3 hours to 6 hours post-LPS, followed by a recovery at 24 hours post-LPS that exceeded baseline levels specifically in ANGPT2OVR but not controltTA mice.
One limitation of our study is that ANGPT2 expression was induced several weeks prior to LPS exposure to facilitate robust elevation in circulating levels. Therefore, this model may not recapitulate the acute release of ANGPT2 from endothelial Weibel-Palade bodies that is presumed to occur in the context of clinical ALI/ARDS, and chronic overexpression of a proangiogenic factor could potentially affect vascular structure and function. However, notwithstanding the possibility of changes in peripheral organs that were not assessed in this lung injury model, we observed no differences in the pulmonary expression of 84 genes involved in vascular biology prior to LPS exposure between ANGPT2OVR and controltTA mice. While not conclusive, this does suggest that the early induction of ANGPT2 under physiological conditions did not significantly alter pulmonary vasculature function, at least at the transcriptional level. Another limitation of this study is that the statistical significance of changes in BAL cell and protein levels assessed at different time points were not adjusted for multiple comparisons; therefore, we cannot discount the possibility of a type I (false positive) result. However, our experiments were designed principally to minimise type II (false negative) results. This is an important pragmatic consideration given limitations with sample size, since breeding double transgenic mice in large numbers represents highly labour-intensive, time-intensive and cost-intensive experiments to conduct. Therefore, we induced elevated plasma levels of ANGPT2 in transgenic mice in an effort to mimic the very high plasma levels observed in patients with ALI/ARDS and used an LPS dose that induced subsaturating effects on pulmonary inflammation and vascular leak (since we hypothesised that high ANGPT2 plasma levels would increase these parameters). In addition, our results showed high internal consistency in the biological effects demonstrated between the various time course, aptamer-mediated ANGPT2 neutralisation and lung transcriptional profiling experiments, and corroborate ANGPT2-mediated proinflammatory44 and anti-leak activities15 that have been independently demonstrated in other laboratories. Thus, while the changes observed in the current study were modest in magnitude, they may still represent biologically meaningful effects. Functional differences between human and mouse ANGPT2 represent another potential limitation associated with the use of the transgenic mouse model in this study. However, human and mouse ANGPT2 are 85% identical in amino acid sequence,9 and previous in vitro 51 and in vivo 15 studies have provided evidence that hANGPT2 is capable of both blocking and inducing the phosphorylation of mouse TIE2 in a context-dependent manner.
In summary, high circulating levels of ANGPT2 exerted only modest effects on pulmonary microvascular function in mice with LPS-induced lung injury, exacerbating inflammatory cell and cytokine infiltration of the alveolar air space while attenuating specific indices of pulmonary vascular leak, with no associated benefit or detriment to survival. Supported by additional transcriptional profiling, these data suggest that elevated circulating levels of ANGPT2 may contribute to vascular homeostatic mechanisms by helping to fine-tune changes in pulmonary gene expression and function in response to proinflammatory stimuli. An important implication is that therapeutic strategies aimed at inhibiting excess circulating levels of ANGPT2 in ALI and sepsis may be less effective than approaches designed to directly activate TIE2, as previously suggested.49 50 This study now broadens the known context-dependent activities of ANGPT2 in vivo, and offers new insight into the possible biological effects of elevated circulating ANGPT2 levels in ALI/ARDS.
Acknowledgments
The authors would like to thank Helen Muleme, Anli Yang, Jack Zhang, Sophie Wen, Mahmoud Salkhordeh and Irene Watpool for technical assistance.
References
Footnotes
Contributors Conception and design: KS and SHJM; data acquisition: KS, MT, YD, SHJM and LM; analysis and interpretation: KS, MT, YD, SHJM and DJS; and manuscript preparation: KS, MT, SHJM and DJS.
Funding This work was supported by the Canadian Institutes of Health Research (MOP82791 to DJS), CREST Ontario Research Excellence Fund (GL2-01-042 to DJS) and Heart & Stroke Foundation of Canada (KS).
Competing interests None declared.
Ethics approval Ottawa Hospital Research Ethics Board.
Provenance and peer review Not commissioned; externally peer reviewed.