Article Text

Abstract
While Pseudomonas aeruginosa (PA) cross-infection is well documented among patients with cystic fibrosis (CF), the equivalent risk among patients with non-CF bronchiectasis (NCFB) is unclear, particularly those managed alongside patients with CF. We performed analysis of PA within a single centre that manages an unsegregated NCFB cohort alongside a segregated CF cohort. We found no evidence of cross-infection between the two cohorts or within the segregated CF cohort. However, within the unsegregated NCFB cohort, evidence of cross-infection was found between three (of 46) patients. While we do not presently advocate any change in the management of our NCFB cohort, longitudinal surveillance is clearly warranted.
- bacterial infection
- bronchiectasis
- cystic Fibrosis
- infection control
- respiratory infection
Statistics from Altmetric.com
Pseudomonas aeruginosa (PA) is a significant pathogen within cystic fibrosis (CF) and non-cystic fibrosis bronchiectasis (NCFB) cohorts. Transmissibility of PA among patients with CF has been widely documented,1 leading to widespread segregation policies.2 In contrast, the cross-infection risk among patients with NCFB is unclear. One UK study concluded that PA cross-infection was rare in NCFB.3 However, in that study, patients with NCFB were managed at a different site from the local CF cohort. Similarly, a recent multicentre study highlighted the potential for PA cross-infection, although this was again exclusively focused on NCFB cohorts.4 In many hospitals, including ours, patients with CF and NCFB share facilities and healthcare professionals. In this context, we conducted a cross-sectional study of PA within our NCFB and CF cohorts to assess the likelihood of cross-infection. In parallel, analysis of local non-respiratory isolates allowed comparison with PA in the wider population.
Sixty-three patients with NCFB and 32 patients with CF were recruited from outpatient clinics based on a documented diagnosis of NCFB/CF and previous PA-positive sputum. PA was subsequently obtained from 46/63 patients with NCFB and 22/32 patients with CF. Ten representative colonies were stored from each PA-positive sputum and were initially genotyped by random amplification of polymorphic DNA (RAPD),5 ahead of multilocus sequence typing (MLST).6 In brief, RAPD was performed on all 10 isolates per patient, and all unique profiles underwent additional evaluation using microfluidic amplicon separation and cluster analysis as described previously.7 This same panel of isolates with unique RAPD profiles was subjected to MLST, enabling strain identification in a global context. Patient demographics and methodologies are detailed in the online supplementary file 1.
Through this approach, 25/46 patients with NCFB (54%) and 13/22 patients with CF (59%) were found to harbour their own unique strain by MLST. The remaining patients harboured strains that were shared within or between cohorts (table 1).
Shared strains of Pseudomonas aeruginosa (PA) identified within the respiratory (CF and NCFB) and non-respiratory cohorts, as defined by MLST. One NCFB patient was coinfected with ST17 and ST564. Isolates in the non-respiratory cohort originated from genitourinary, wound, ENT and faecal samples from community and hospital investigation
All shared strains within our respiratory cohorts are globally distributed according to the MLST database, and the majority have been isolated from diverse clinical and environmental sources. Consistent with this, we observed many of the same strains within our non-respiratory cohort (table 1). Given the ubiquitous nature of these strains, their presence in multiple patients may reflect independent acquisition rather than cross-infection, and neither RAPD nor MLST provide sufficient resolution to address this. Consequently, whole genome sequencing (WGS) was performed on the shared strains from the respiratory cohorts to assess relatedness at a whole-genome level. For ST17 (the most prevalent strain observed), three isolates per patient were sequenced to enable assessment of inter-patient and intra-patient diversity. For all other shared strains, one isolate was sequenced per patient.
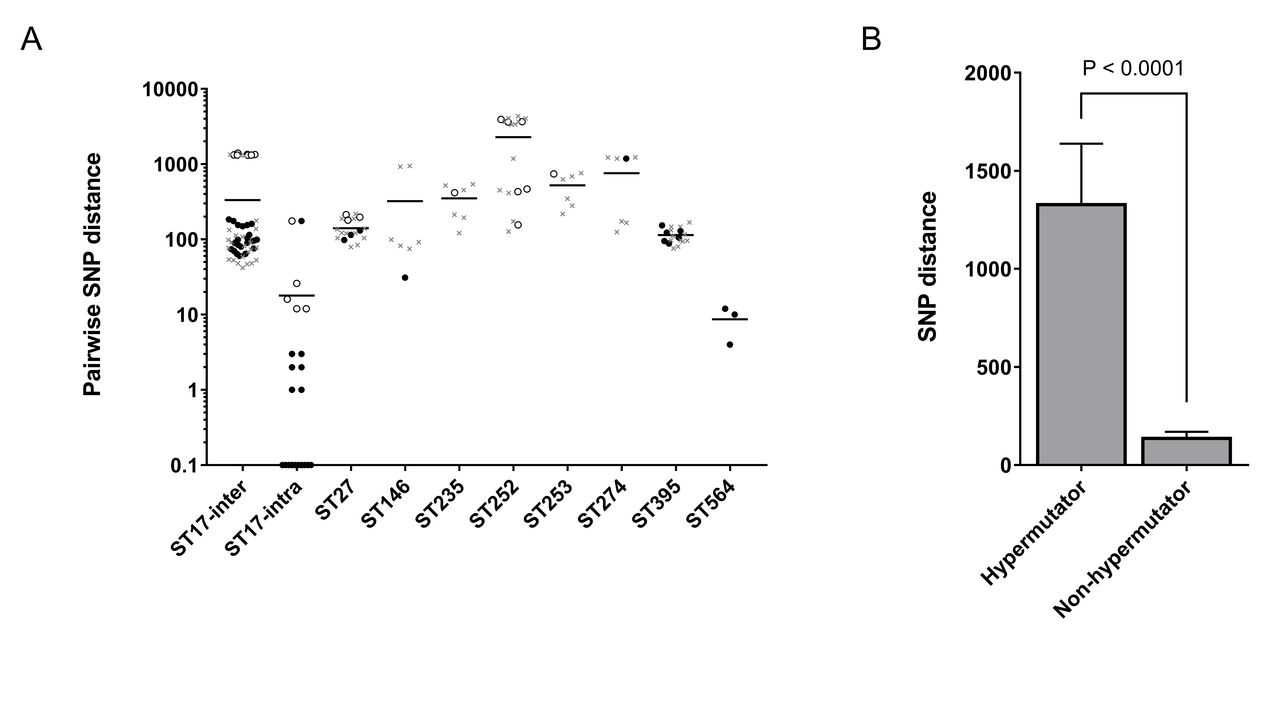
In WGS-based studies, patient-to-patient transmission cannot be defined based on a simple threshold of the number of single nucleotide polymorphisms (SNPs) between isolates. Hypermutation accelerates genetic divergence, and hypermutable PA are commonly observed within chronic lung infections.8 Consistent with this, in silico prediction of hypermutators revealed putative hypermutable PA within our respiratory cohorts (see online supplementary file 1) and predicted hypermutator status correlated strongly with SNP distance (figure 1B), highlighting the difficulty in setting a SNP threshold. Consequently, to aid interpretation, we incorporated relevant publicly available genome sequences into our WGS-based analysis, enabling us to compare PA isolates from our respiratory cohorts with representative PA that belong to the same sequence type but are not epidemiologically linked to our cohorts (see online supplementary file 1). Furthermore, when considering likelihood of cross-infection, relevant patient-specific and strain-specific information was reviewed, including potential cross-infection events, duration of infection, change in culture status and knowledge of strain distribution and transmissibility.
{kind=link}
Genetic diversity within Pseudomonas aeruginosa (PA) isolates, as defined by whole genome sequencing. (A) The number of single nucleotide polymorphisms (SNPs) was calculated across the core genome of all sequenced isolates. Each data point represents a pairwise comparison within each ST, with the bar representing the mean. Circles represent pairwise comparisons that are exclusively between PA isolates from our own respiratory cohort (cystic fibrosis or non-cystic fibrosis bronchiectasis), with the open circles representing those comparisons in which at least one isolate is a predicted hypermutator. The grey crosses represent pairwise comparisons in which one isolate is from our respiratory cohort and the other is an unconnected representative of the same sequence type (using publicly available genomes). For ST17, SNP numbers are shown that reflect the diversity observed between patients (ST17-inter) and within individual patients (ST17-intra; based on sequencing of three isolates per patient). (B) Predicted hypermutable PA isolates exhibited significantly elevated levels of genetic divergence (SNP distance) relative to predicted non-hypermutable PA.
The WGS analysis revealed that the CF and NCFB isolates belonging to ST17, ST27, ST235, ST252, ST253, ST274 and ST395 are as divergent from each other (figure 1A; circles) as they are from unconnected representatives of the same sequence type (figure 1A; grey crosses). Furthermore, with the exception of two intra-patient pairwise comparisons (one involving a predicted hypermutator), analysis of the ST17 isolates revealed significantly greater ST17 diversity between patients than within patients. While cross-infection cannot be completely ruled out, particularly for the divergent ST17 group, we conclude (on the basis of inter-patient diversity, the ubiquitous nature of these strains and a review of relevant clinical information) that the occurrence of these seven sequence types in multiple patients most likely reflects independent acquisition.
In contrast, two shared strains exhibited low inter-patient diversity that we believe to be indicative of cross-infection. First, two CF isolates of ST146 (Liverpool epidemic strain (LES)) differed by only 31 SNPs and were more closely related to each other than to unconnected representatives of the LES (figure 1A). The two patients involved were siblings who had become colonised with PA before coming under the care of our unit. The genetic relatedness of the isolates coupled with the high level of personal contact between patients and the known transmissibility of LES strongly supports cross-infection.
More significantly, the ST564 isolates from three unrelated patients with NCFB are near-identical, differing by only 4–12 SNPs. While no publicly available genomes of ST564 representatives were available for comparison, we believe this extremely high level of genetic relatedness is indicative of cross-infection, a conclusion further supported by clinical records that revealed two of the three patients shared a waiting area and lung function room approximately 17 months prior to recruitment. This potential cross-infection event did not coincide with a clear change in PA culture status as one of the patients intermittently isolated PA before and after this event, while the other patient had evidence of multiple PA strains (and therefore superinfection may have occurred). While we were unable to identify potential cross-infection event(s) involving the third ST564-infected patient (who also carried multiple strains), a difference of only four SNPs strongly supports cross-infection. Interactions may have occurred in or outside the hospital that are not apparent via the review of clinical notes. Furthermore, we believe ST564 acquisition from a common environmental source is highly unlikely due to its absence from other cohorts.
In agreement with previous literature,3 but expanding it to high-resolution WGS analysis, we therefore conclude that PA cross-infection is highly likely to have occurred within our NCFB cohort. While we believe this to be restricted to ST564, additional cross-infection events involving other sequence types cannot be definitively ruled out, particularly given the confounding role of hypermutators. Similarly, on the balance of evidence, we consider it unlikely that cross-infection has occurred between CF and NCFB cohorts despite them sharing facilities, and notable differences in strain distribution between the cohorts argue against the presence of an environmental reservoir within the unit. Our studies suggest that ST564 has the potential for transmissibility and superinfection. Although not reported in the literature, the MLST database (https://pubmlst.org/paeruginosa 9) reports ST564 as having been isolated from sputum (Netherlands) and water (Australia and France).
At present, we believe the negative impacts that would be associated with implementing a segregated NCFB cohort (including reduced patients per clinic and reduced access to pulmonary rehabilitation courses) outweigh the low risk of cross-infection. However, with growing NCFB cohorts nationwide10 and cross-infection possible, ongoing longitudinal surveillance is clearly warranted.
Acknowledgments
The authors would like to thank all the patients who participated in this study; Hilary Mortimer, Sarah Murray, Rachel Rogers, Jayne Trott, Roseanne Lowless and Miriam Green for their help in obtaining samples; the Microbiology Department at the Royal Devon and Exeter Hospital, particularly Nigel Richardson and Dr Cressida Auckland and Dr Matthew Robinson and Audrey Farbos for laboratory guidance. The authors acknowledge the use of the University of Exeter’s Zeus computational infrastructure for analysis of whole genome sequence data. The project was supported by NIHR Exeter Clinical Research Facility, and the authors thank Chloe Slade and Kathryn Shears for their assistance in managing samples and data.
Footnotes
Contributors PJM, CDS, NJW and ARB: study concept and design. PJM, CDS and NJW: patient recruitment. PJM, JR and KAM: methodology and investigation. PJM, MJB, PAO, KP, EM and ARB: data analysis and interpretation. PJM and ARB: writing the original draft. PJM, MJB, EM, CJS, CDS, NJW and ARB: writing, reviewing and editing the manuscript.
Funding This study was funded though charitable donations and a Small Grants Award by the Research and Development department at the Royal Devon and Exeter NHS Foundation Trust. The funding sources had no input into this study or its submission for publication.
Competing interests EM declares a grant from AlgiPharma AS held as a service contract on a CF clinical trial (unrelated to this work).
Patient consent Obtained.
Ethics approval Ethical approval for the study of our NCFB cohort was obtained through the NRES Committee South West- Exeter (14/SW/0080). Our CF samples and data were collected through the RD&E tissue bank (11/SW/0018).
Provenance and peer review Not commissioned; externally peer reviewed.