Article Text

Abstract
The microbiome has been proposed to play a role in the progression of idiopathic pulmonary fibrosis (IPF) based on bronchoalveolar lavage analyses, but the microbiome of lung tissue in IPF has not been explored. In a case–control study of lung explants analysed by 16S rRNA gene sequencing, we could not reliably detect bacterial DNA in basilar tissue samples from patients with either chronic or acute exacerbations of IPF, in contrast to control candidate-donor lungs or cystic fibrosis explants. Thus, our data do not indicate microbiome alterations in regions of IPF lung with advanced fibrosis.
- interstitial fibrosis
- respiratory infection
- bacterial infection
Statistics from Altmetric.com
Introduction
In the quest for environmental factors leading to the development and progression of idiopathic pulmonary fibrosis (IPF), recent research has implicated the lung microbiome.1 The microbiome hypothesis was generated by clinical observations of the impact of interventions with plausible effects on lung microbiota, such as antibiotics, immunosuppression and gastric acid suppression.1 Direct supportive evidence came from two well-designed studies using next-generation sequencing to analyse bronchoalveolar lavages (BALs) from early-stage IPF patients. While these two studies implicated the baseline microbial load2 or specific taxa composition3 in IPF progression and associated host responses,4 5 we currently lack information about the microbiome in lung tissue or in end-stage IPF. Furthermore, objective data on dysbiosis during acute exacerbations (AEIPF) are limited despite conjectures about microbial triggers.6 For these reasons, we conducted the Microbiome in Lung Explants study in IPF (MiLEs-IPF) to define discriminating features of the lung tissue microbiome in IPF and potential microbial perturbations in AEIPF.
Methods
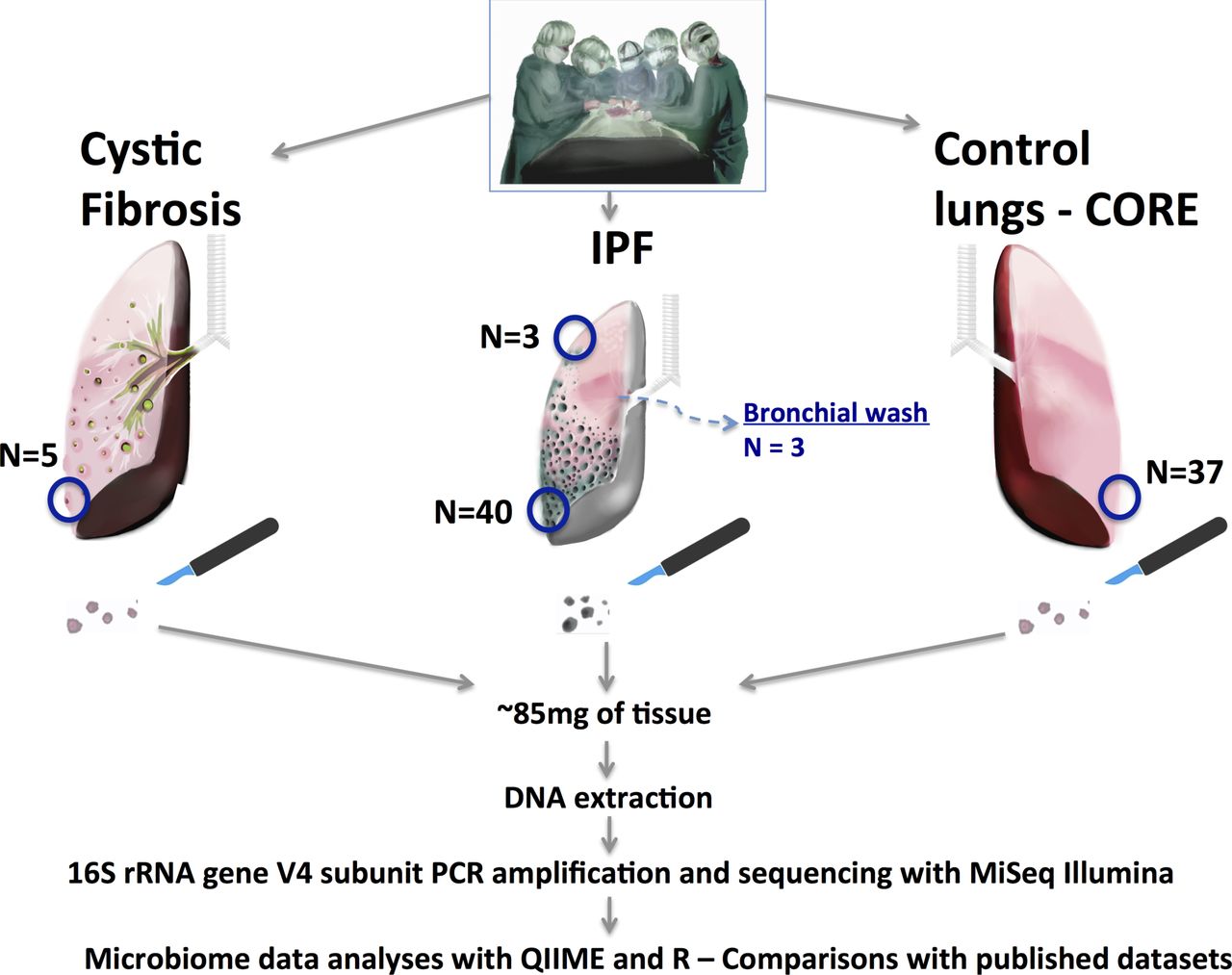
See online supplementary material for detailed methods. Briefly, MiLES-IPF is a case–control study comparing the microbiome composition of tissue specimens taken from lung explants (figure 1). We resected subpleural lower lobe tissue samples from explants from patients with end-stage IPF (n=40) at the time of lung transplantation or post mortem. We obtained 37 control tissue samples from lung donation candidates deemed unsuitable for transplant via the Center for Organ Recovery and Education (CORE). We also included five controls from patients with cystic fibrosis (CF), a disease with high microbial load in lung tissue. Microbiome experiments and analyses were performed according to well-established protocols and analytical pipelines (online supplementary material).
Supplementary file 1

Design of the MiLES-IPF study. We performed a case–control study with collection of IPF lung tissue samples in the operating room (for lung transplant cases) or the morgue (for patients undergoing rapid autopsy). We included lung explants from 40 patients with IPF: 10 with acute exacerbations, 31 men (78%), mean age 63 years and mean predicted forced vital capacity 46%. Similar processes were performed for control donor lungs (CORE—32 organ donation candidates offering 37 tissue samples) and five cystic fibrosis explants. Under sterile conditions, we resected basilar, subpleural pieces from each lung (weighing ~85 mg on average). In small subsets of patients with IPF, we also obtained matched apical (or middle lobe) tissue samples and performed bronchial washings immediately post-explantation for analysis of the lavage fluid. Tissue samples were flash frozen to −80°C until experiments with DNA extraction, PCR amplification of the hypervariable V4 region of the highly conserved 16S rRNA gene and sequencing with the Illumina MiSeq platform. CORE, Center for Organ Recovery and Education; IPF, idiopathic pulmonary fibrosis; QIIME, Quantitative insights into Microbial Ecology.
Results
Of 40 IPF patients, 10 with AEIPF had similar demographic and pulmonary function test characteristics as those with chronic IPF (see online supplementary table S1), but higher prevalence of diffuse alveolar damage on pathology (p<0.05). To infer relative bacterial loads, we examined the number of high-quality 16S sequences (reads) produced by sequencing of each tissue sample (figure 2A). IPF lungs (both AEIPF and chronic) had an exceedingly low mean number of reads (183 (244)) in the range of reagent negative controls, a yield that was at least 15-fold smaller compared with CORE or CF lungs (p<10−5 each). Similarly, by quantitative PCR, we were unable to detect any quantifiable signal of 16S rRNA gene copies in all IPF lung samples, whereas we did so in 36% of CORE or CF samples (figure 2C).

{kind=link}
{kind=link}
Main results of microbiome analyses in MiLES-IPF. (A) Comparison of number of 16S reads between different types of samples. IPF lungs had much lower number of reads compared with CORE and CF lungs (p values <10−5). (B) Principal coordinates analysis of Bray-Curtis dissimilarity distances between different types of samples. Higher distances between samples (depicted by spheres) indicate higher levels of taxonomic dissimilarities. (C) Quantitative PCR (qPCR) of the 16S rRNA gene results with end-point fluorescence (EPF) shown (y axis) for each sample (x axis). Samples are grouped according to types (CF, CORE, IPF, negative controls, positive controls). Each bar indicates a different sample. The cut-off EPF for quantitation of 16S rRNA gene signal is shown with a dashed line. Given that only 36% of CF and CORE samples provided quantifiable 16S qPCR signal despite having high numbers of 16S reads by sequencing (as shown in panel A), the microbial mass present in our tissue samples had to be at the limit of detection of the qPCR protocol (see online supplementary material) ; however, no IPF or negative control sample produced a quantifiable 16S qPCR signal. (D) Taxonomic composition of individual samples classified by sample type. Each bar represents a patient sample and relative heights of each colour-coded bar indicate relative abundance of corresponding taxa. Cystic fibrosis-related pathogens are coded in red, common oral taxa are coded with variations of blue and skin taxa are coded with variations of brown. CORE, Centre for Organ Recovery and Education; IPF, idiopathic pulmonary fibrosis; CF, cystic fibrosis.
Further ecological analyses suggested that the trace microbial signal in IPF samples was accounted for by experimental contamination (background signal). By principal coordinates analysis of Bray-Curtis dissimilarity distances (figure 2B), IPF samples clustered with negative reagent controls and were statistically dissimilar from CORE samples (anosim p<0.001). CF communities were dominated by Burkholderia or Pseudomonas taxa (corresponding to pathogens identified in explant cultures) and CORE samples had overall higher abundance of typical oral taxa (eg, Prevotella, Streptococcus) as expected for the supraglottic pneumotype of the healthy lung microbiome.7 In contrast, IPF samples had high abundance of typical skin microbiota (eg, Comamonadaceae, Methylobacterium) also seen in negative controls (figure 2D). Statistically significant differences in relative abundance (adjusted for multiple comparisons) were found for 33 taxa between IPF and CORE lungs (online supplementary figure S3), and bioinformatics analyses confirmed differential abundances of ‘oral’ and ‘skin’ origin taxa in IPF and CORE lungs (online supplementary figure S4). AEIPF was taxonomically indistinct from chronic IPF (online supplementary figure S5).
To examine for possible spatial heterogeneity of microbial communities in IPF lungs, we compared matched apical and basilar samples for a small available subset of patients (n=3), but found no consistent differences in microbial load or taxonomy (online supplementary figure S7). To assess for potential differences between airway-based and tissue-based samples, we compared another subset of matched post-explant bronchial washings and tissue samples, and identified overall higher number of reads in bronchial washings but limited taxonomic overlap with tissue samples (range of overlapping taxa 29% to 53%) (online supplementary figure S8). As expected, our tissue-based IPF samples had entirely discordant taxonomic composition compared with BAL IPF samples from a previous study (online supplementary figure S9).2
Discussion
Our analyses in a well-phenotyped cohort of lung explants from end-stage IPF patients provided a surprisingly low bacterial signal that was similar to negative control samples. Furthermore, no differences were detected between AEIPF and chronic IPF. Technical reasons are unlikely to account for these null results: the low signal in IPF samples was consistent among replicated experiments, and in striking contrast with the strong microbial signal from CF lung explants, where we detected high abundance of clinically identified pathogens, and from CORE lungs, which demonstrated features of the supraglottic phenotype of the normal lung microbiome in most cases. Due to our sampling strategy targeting basilar, subpleural lung regions with advanced honeycombing, we may have encountered ‘microbiome deserts’ inhospitable for bacterial growth. Furthermore, the hallmark temporal heterogeneity of usual interstitial pneumonia may create spatial heterogeneity of host–microbiota interactions along the apico-basilar gradient of fibrosis; however, we were unable to detect any substantial differences in a small subset of three patients with matched apical–basilar samples.
Notably, our findings in honeycombed tissue of end-stage IPF differ from previous BAL-based studies in early IPF.2–5 The observed discordances are likely attributable to sampling strategy differences, that is, whether the microbiome is examined starting from the airways or the pleura (online supplementary figures S8 and S9). BAL sampling probably captures wider areas of alveoli and small airways compared with our tissue samples. It is possible that the resident microbiota in IPF lungs demonstrated in previous studies arrive in the respiratory tract by micro-aspiration associated with IPF,1 8 and then they remain predominantly in the bronchiectatic airways with impaired clearance and not in the distal fibrosed parenchyma.
Despite methodological limitations of our study (ie, cross-sectional design, single sample per explanted lung, unavailability of strictly normal tissue, small sample size), the relevant finding is that parenchymal samples with advanced IPF had effectively no detectable microbiota, in contrast with other types of explant tissue. Our negative findings do not invalidate the microbiome hypothesis in IPF, but make a call for topographically detailed investigations on the spatial heterogeneity and possible airway predominance of host–microbiota interactions in IPF.
Acknowledgments
The authors would like to thank Mr Evaggelos Makkas for the graphic design of figure 1.
Footnotes
Contributors Study conception and design: GDK, MR, JCS, JM, BJM, AM. Data acquisition: GDK, MR, AF, JCS, SQ, KLV, DJK, KL, KFG, JMP, BM, KL, BJM, JM, AM. Data analysis and interpretation: GDK, MR, AF, JCS, SQ, KLV, DJK, KL, KFG, BM, JMP, KL, BJM, JM, AM. Drafting of the manuscript: GDK, MR, AF, JCS, DJK, KLV, BJM, AM. Critical revision of the manuscript: GDK, MR, AF, JCS, SQ, KLV, DJK, KL, KFG, BM, JMP, KL, BJM, JM, AM. Approval of manuscript for submission: GDK, MR, AF, JCS, SQ, KLV, DJK, KL, KFG, BM, JMP, KL, BJM, JM, AM.
Funding National Institutes of Health T32 HL007563-29, K24 HL123342, R01 HL123766-01A1, P30 DK 72506, Breathe Pennsylvania Lung Health Research Grant and Cystic Fibrosis Foundation Research Development Program, Center for Medicine and the Microbiome University of Pittsburgh.
Competing interests None declared.
Ethics approval The University of Pittsburgh Institutional Review Board and Committee for Oversight of Research and Clinical Training Involving Decedents approved the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Research letter
- Airwaves