Article Text

Statistics from Altmetric.com
Case report
A 22-year-old woman was referred to our respiratory outpatient clinic to undergo a cardiopulmonary exercise test (CPET), following presentation with a 2-year history of progressive unexplained exertional dyspnoea (ED). Specifically, she described dyspnoea precipitated by walking or running, which gradually improved with rest. She denied chest pain, cough, asthenia or muscular weakness. Until the age of 20, she practised regular non-competitive physical activity.
The patient had at term natural childbirth and normal psychophysical development. Menstrual period, started at the age of 14 years, was regular; no pregnancy, nor miscarriage. She was a lifelong non-smoker with no history of illicit substance use or occupational exposure (university student). Her past history included multinodular euthyroid goitre for which she was prescribed regular levothyroxine (75 μg/100 μg on alternate days). She also reported intermittent gastrointestinal discomfort, which had been ascribed to lactose intolerance for which, however, she was not prescribed a lactose-free diet. Her family history revealed an uncle deceased during childhood for ‘cardiac arrhythmia’.
Cardiorespiratory and neurological examination was unremarkable. She was normotensive (blood pressure (BP) 100/60 mm Hg, heart rate (HR) 98 bpm in sinus rhythm), not tachypnoeic (respiratory rate 16 breaths/min) and had preserved oxygen saturation (SatO2 98%). Her body mass index was 19.3.
Pulmonary function tests showed normal flow indices (FVC 3.52 L—88% of pred; FEV1 3.35 L—96% of pred; Tiffeneau index FEV1/FVC 0.95) and preserved lung volumes and gas transfer (total lung capacity 3.94 L—90% of pred; diffusing capacity of the lungs for carbon monoxide, 96% of pred). A high-resolution chest tomography showed no sign of lung disease.
Cardiological (including rest, stress and Holter ECG, as well as cardiac ultrasounds) and neurological (including electromyography) exams showed no abnormalities.
Routine blood tests (cell count, glucose and electrolyte levels, iron and ferritin profile, lipid screening, kidney and liver function), erythrocyte sedimentation rate, C reactive protein and muscular enzyme (creatine kinase with isoenzymes) values were within normal ranges. Thyroid hormones (measured during replacement therapy) were normal: thyroid-stimulating hormone (TSH) 0.31 µUI/mL, FT3 2.94 pmol/L, FT4 13.8 pmol/L. Autoimmune screening, including antinuclear antibodies, extractable nuclear antigens, anti-double stranded DNA antibodies, anti-β2 glycoprotein I antibodies, anticardiolipin antibodies, lupus anticoagulant and antineutrophil cytoplasmic antibodies (pANCa, cANCA), was negative.
An incremental load (10 W/min) maximal (peak respiratory exchange ratio (RER) 1.6) CPET was conducted on a cycle ergometer till exhaustion for prevalent muscular fatigue (10-point Borg scale for rating of perceived exertion (RPE)1 at peak exercise: dyspnoea 9/10, leg fatigue 10/10), in the absence of cardiorespiratory symptoms (table 1).
Baseline compared with follow-up cardiopulmonary exercise test (CPET) parameters
The test showed a severe reduction in exercise capacity (VO2peak 704 mL O2/min or 12.8 mL O2/min/kg, 38% pred) with impaired maximal workload (60 W, 35% pred) and work efficiency (VO2/W slope 5.4 L/W/min, 52% pred).
The cardiac response revealed high resting hearth rate (110 bpm), with a rapid depletion of the HR reserve (176 bpm, 89% of max HR pred at 60 W) and a low O2 pulsepeak value (4.0 mL O2/bpm, 40% pred). Values of BP were within normal ranges both at rest and at peak of exercise. No arrhythmias, ventricular repolarisation abnormalities and signs of myocardial ischaemia were seen.
The analysis of the respiratory pattern during exercise revealed impaired ventilatory efficiency (VE/VCO2LT 42, VE/VCO2slope 39) with normal VEpeak value (46.8 L/min, 38% of maximum voluntary ventilation (MVV)) and preserved breathing reserve at exercise cessation with no significant desaturation. Of notice, ventilatory equivalents for O2 at peak exercise reached very high values (VE/VO2peak 65). At peak exercise, end-tidal oxygen tension (PetO2) was high (127 mm Hg), while end-tidal carbon dioxide tension (PetCO2) was low (29 mm Hg).
The study of the metabolic profile was determined using combined assessment of ‘V-slope’,‘ventilatory equivalents’ and ‘end-tidal pressure’ methods and highlighted an early lactic threshold (27% pred; normal values >35%).
Overall, the reduced CPET exercise tolerance, in the presence of a progressive rise of RER throughout exercise and a reduced anaerobic threshold (AT), suggested early onset of lactic acidosis. Observed high VE/VCO2 and low PETCO2 values might relate to increased activation of metabolic chemoreceptors within skeletal muscle; however, the exact underlying mechanism has not been yet fully elucidated. Furthermore, the abnormalities of O2 pulse and the markedly impaired VO2/W slope were indicative of a severe defect in O2 delivery and/or muscle O2 extraction.
Arterial blood gas (ABG) sampling, breathing room air, showed a mixed acid–base disorder at rest (pO2 111 mm Hg, pCO2 35 mm Hg, pH 7.40, HCO3- 21 mmol/L, lactate 2.2 mmol/L) and a significant metabolic acidosis at peak exercise (pO2 118 mm Hg, pCO2 26 mm Hg, pH 7.19, HCO3- 9.9 mmol/L, lactate >20 mmol/L).
The above clinical and laboratory findings, as well as the CPET and ABG parameters, in the absence of pulmonary and cardiovascular diseases, were in favour of a diagnosis of a metabolic disorder characterised by altered skeletal muscle oxygen use. A muscular biopsy was therefore performed. According to the protocol adopted in our laboratory, the bioptic sample underwent a panel of histological, histochemical and immunohistochemical analyses, including H&E, Gomori trichrome, sequential cytochrome c oxidase/succinate dehydrogenase (COX/SDH) and ATPase stains. Immunohistochemical analyses with the following antibodies were also performed: fast, slow and neonatal myosin, dystrophin (N-terminus, rod domain, C-terminus), sarcoglycans (α, β, δ, γ), dysferlin, major histocompatibility complex class I, CD68. At histological analysis, the most peculiar feature was the presence of several fibres with increased subsarcolemmal intensity at COX/SDH stain, compatible with mitochondrial proliferation (figure 1A, B). There was only one COX-deficient fibre (characterised by a slight stain reduction), although COX-negative fibres were absent. Gomori trichrome stain did not show ragged red fibres. In addition, there was a slight variability in the fibre size, without muscle necrosis or degeneration (figure 1A, B). There were neither inflammatory infiltrates, nor endomysial fibrosis. Mitochondrial subsarcolemmal proliferation was confirmed by ultrastructural examination, which did not reveal cristae alterations and paracrystalline inclusions (data not shown). These findings suggested a diagnosis of a mitochondrial myopathy.

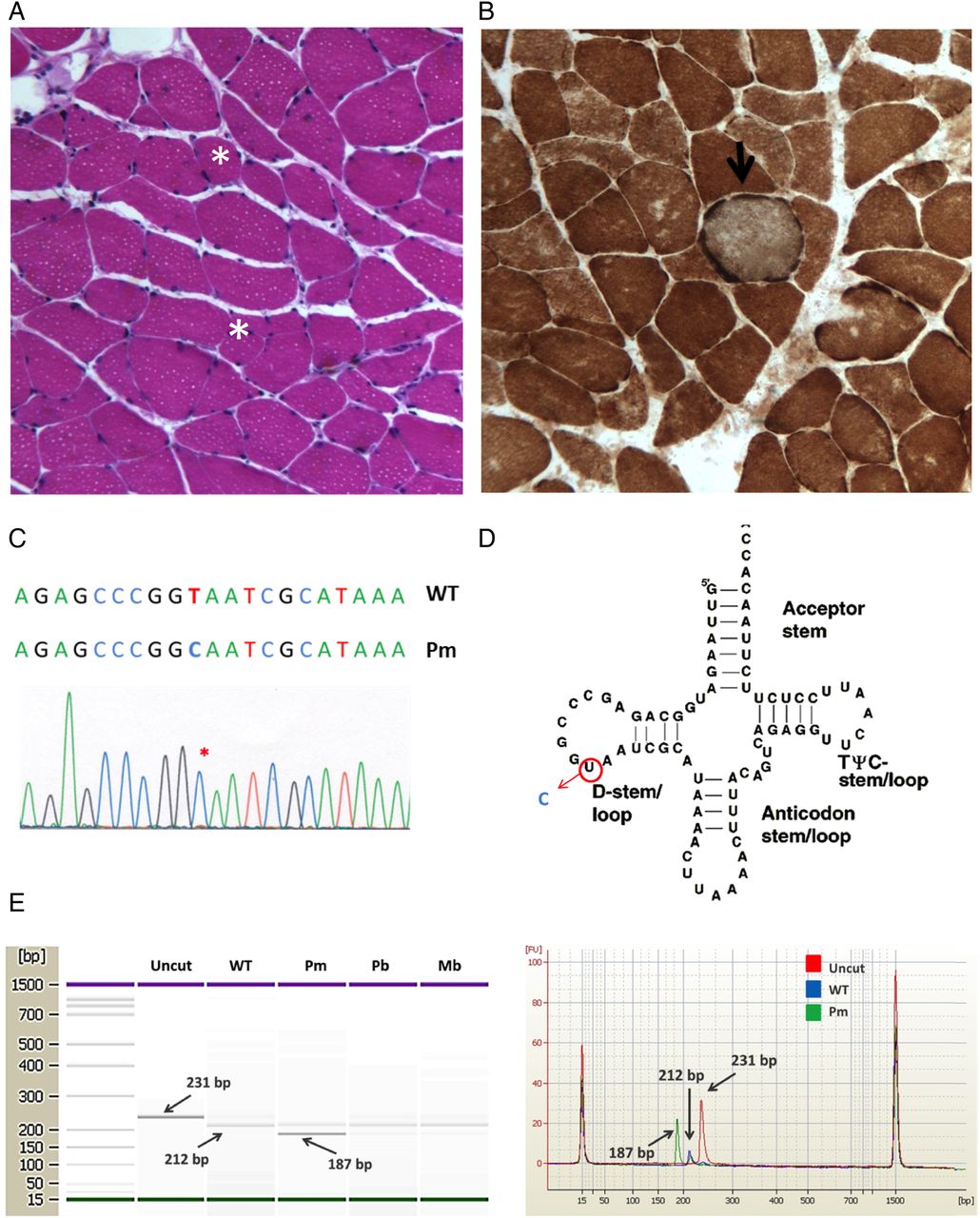{kind=link}
Identification of the m.3250T>C mutation. (A) Histological analysis of the muscle biopsy from the proband showing only slight variation in fibre size (asterisk shows ipotrophic fibres; H&E, original magnification ×20). (B) Sequential cytochrome c oxidase (COX)/SDH histochemistry on skeletal muscle biopsy from the proband showing a COX-positive muscle fibre with increased subsarcolemmal staining (arrow), compatible with mitochondrial proliferation (original magnification ×20). (C) Sequence electropherogram of mt-tRNALeu(UUR) showing the m.3250T>C mutation in the proband's skeletal muscle (mutation is indicated by an asterisk). (D) Secondary structure of mt-tRNALeu(UUR) with the position of the m.3250T>C mutation highlighted in the D-loop. (E) Restriction fragment length polymorphism analysis of 3250T>C mutation. The restriction endonuclease NaeI cleaves wild-type sequences (size 231 bps) in two fragments (sized 212 and 19 bps; the latter is not visible in the figure). The mutation introduces a second NaeI recognition site that cleaves the 212 bps in two smaller fragments (sized 187 and 25 bps; the latter is not visible in the figure). Restriction fragments were separated using the Agilent 2100 bioanalyzer instrument with the 1000 LabChip kit. Left: LabChip gel image. Right: overlay of three different electropherograms (red=uncut; blue=wild type; green=3250T>C mutant). The bands are separated and identified by the software as separate peaks. WT, wild type; Pm, proband skeletal muscle; Pb, proband blood; Mb, =proband mother's blood.
To confirm the diagnosis, direct sequencing of whole mitochondrial DNA (mtDNA) from the muscle homogenate was performed, revealing the m.3250T>C mutation in the MTTL1 gene (figure 1C, D). Identical change was found in mtDNA from peripheral lymphocytes of the patient and of her mother, although clinically unaffected. Restriction fragment length polymorphism analysis showed that the m.3250T>C mutation was heteroplasmic, with a mutation load of ∼80% in the proband's muscle, ∼20% in the proband's blood and ∼10% in the mother's blood (figure 1E).
On the basis of the literature data, supplemental therapy with L-carnitine (5 g/day) and riboflavin (200 mg/day) was started, and the patient was asked to return for a 1-year reassessment.
At the follow-up visit, the patient reported a significant improvement in exercise capacity. A repeat maximal (RER peak 1.6) CPET (table 1), performed according to the same exercise protocol, continued to demonstrate impaired exercise capacity (VO2peak 14.6 mL O2/min/kg, 41% pred), low maximal work rate (60 W, 35% pred), early lactate threshold (30% pred), low O2 pulsepeak value (4.6 mL O2/bpm, 47% pred), mildly impaired ventilatory efficiency (VE/VCO2slope 30) and high ventilatory equivalents for O2 (VE/VO2peak 56). However, resting HR was now within normal ranges (86 bpm) with a preserved heart rate reserve (HRR) (162 bpm, 82% of max HR pred at peak exercise). Borg scale scores for RPE were also significantly improved (at peak exercise: dyspnoea 4/10, leg fatigue 5/10). Peak exercise ABG sampling demonstrated significant improvement in peak exercise lactate: pH 7.27, HCO3- 18 mmol/L, lactate 8.7 mmol/L.
Discussion
ED is a common complaint in young people. The possible underlying causes are numerous, including a wide range of pulmonary and cardiovascular abnormalities. Metabolic causes are however often overlooked as a potential aetiology, and in particular, inborn errors of metabolism or mitochondrial disorders can remain unidentified, especially when presenting in a mild form. Such patients, in fact, may have normal pulmonary and cardiac exams, often misleading to a wrong diagnosis of ‘psychogenic exertional dyspnoea’.
For patients with unexplained dyspnoea and for whom test results at rest are non-diagnostic, there is robust evidence suggesting that CPET represents a valuable diagnostic tool.2 In fact, CPET is able to provide an integrated and accurate evaluation of the cardiovascular, respiratory and metabolic responses to workloads, as well as to estimate potential pathological conditions in the three compartments, affecting exercise capacity.3 Furthermore, results from CPET may efficiently direct further diagnostic testing to target the suspected organ or system involved. On the other hand, normal CPET findings limit subsequent time-consuming, invasive and expensive investigations. CPET also allows assessing and monitoring along time the individual's exercise tolerance, which is widely recognised as one of the strongest endpoints in the estimate of the quality and expectancy of life, in both health and disease.4 At last, peak exercise blood gas sampling can detect an ongoing metabolic acidosis state that is clearly evident only at high workloads, providing a direct measurement of arterial blood lactate levels.5
In our case report, we observed a reduced CPET exercise tolerance combined with early anaerobic threshold, altered ventilatory and work efficiency and impaired oxygen pulse and end-tidal carbon dioxide pressure. In the presence of raised lactate levels both at rest and, particularly, at peak exercise, and in the absence of cardiopulmonary, blood, kidney and liver abnormal findings, as well as of any history of alcohol or drug poisoning, a metabolic disorder, characterised by altered skeletal muscle oxygen use, was mainly suspected. The evidence of a primary mitochondrial disease was then obtained through the muscle biopsy and the mtDNA sequencing.
The term primary mitochondrial diseases is typically used to describe a heterogeneous group of systemic disorders in which energy production is impaired through the alteration of mitochondrial oxidative phosphorylation. Mitochondriopathies in children have a prevalence of approximately 1 in 10,000. Among adults, it has been reported a prevalence of 1 in 5000 individuals, despite carriage of pathogenic mtDNA mutations may be found at significantly higher rates of up to 1 out of 200.6 Symptoms are largely heterogeneous within the course of the disease and may be extremely severe and even fatal. Milder forms, reported in literature, can however present with non-specific clinical symptoms and signs, leading patients to seek medical advice for chronic fatigue, muscle pain or ED. Of the approximately 80 polypeptides that constitute the five complexes of the respiratory chain, 13 are encoded by the mitochondrial DNA (mtDNA), while the others by nuclear DNA (nDNA). Therefore, mitochondrial diseases can be caused by mutations in either mtDNA or nDNA. Disorders due to mutations in mtDNA are transmitted by maternal inheritance, whereas disorders due to mutations in nDNA are transmitted by classic Mendelian inheritance. During the last two decades, up to 600 (http://www.mitomap.org) pathogenic mutations of mtDNA have been associated with multiple syndromes, mainly characterised by a multisystemic involvement. Most of the known mtDNA mutations affect the transfer RNA (tRNA) genes, impairing the overall synthesis of mitochondrial proteins and causing massive proliferation of mitochondria in skeletal muscle.
Our case report showed a T-to-C transition at nucleotide 3250 in the mtDNA–tRNALeu(UUR) gene (MTTL1). Mutation involving this gene has been reported only twice in literature7 ,8: the first report described a family with several members affected by chronic fatigue and muscular weakness, with five siblings who died in early childhood of unknown causes; the second described a female patient with muscular weakness onset in early childhood, with two cases of unexplained infant death in her family.
Even though the majority of mitochondrial disorders have no specific treatment,9 ,10 it has been reported that some forms may benefit from supplementation of carnitine, riboflavin and coenzyme Q10, as well as from programmes of physical training, leading to better exercise tolerance and to an improvement of symptoms.8 ,11–13 It has been in fact suggested that the use of these precursors of coenzymes may result in a clinical response by stimulation of enzyme activity.14 Riboflavin showed a beneficial effect in several patients with Complex I deficiency.15 ,16 In three of those patients, a defect in Complex I activity was associated with normal carnitine levels in serum but decreased carnitine content in muscle, while four had low serum carnitine levels, suggesting the need of implementation.17–19
Our patient reported a significant clinical improvement with reduced respiratory discomfort during exercise after 1 year of riboflavin and carnitine supplementation. Furthermore, at the 12-month follow-up visit, the patient showed a slightly increased VO2max (14.6 vs 12.8 mL O2/min/kg), improved chronotropic competence (HRrest 86 vs 110 bpm; HRpeak 162 vs 176 bpm) and ventilatory efficiency (VE/VCO2slope 30 vs 39), with a lower arterial lactate value (8.7 vs >20 mmol/L).
Conclusions
We reported a case of ED due to a very rare mutation (m.3250T>C) in the MT-TL1 gene. CPET, integrated with blood gas sampling at peak exercise, revealed a useful diagnostic tool to unmask a mitochondrial myopathy in our patient and to monitor its course. The subjective and objective improvements observed with a chronic riboflavin and L-carnitine supplementation therapy should call for controlled studies on the potential efficacy of this treatment strategy in patients with mitochondrial diseases.
Acknowledgments
The authors thank Professor Giulia d'Amati and Dr Carla Giordano for providing the histological diagnosis on muscle biopsy and the results of mutation analysis on blood and tissues.
References
Footnotes
Contributors MI, MB and PM performed clinical, functional and laboratory assessments. EP and BC performed the muscle biopsy and the related histological, histochemical and immunohistochemical analyses. All authors significantly contributed to data interpretation and manuscript preparation.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.