Article Text

Abstract
Introduction Non-small cell lung cancer (NSCLC) is a heterogeneous disorder consisting of distinct molecular subtypes each characterised by specific genetic and epigenetic profiles. Here, we aimed to identify novel NSCLC subtypes based on genome-wide methylation data, assess their relationship with smoking behaviour, age, COPD, emphysema and tumour histopathology, and identify the molecular pathways underlying each subtype.
Methods Methylation profiling was performed on 49 pairs of tumour and adjacent lung tissue using Illumina 450 K arrays. Transcriptome sequencing was performed using Illumina HiSeq2000 and validated using expression data from The Cancer Genome Atlas (TCGA). Tumour immune cell infiltration was investigated by immunohistochemistry.
Results Unsupervised hierarchical clustering of tumour methylation data revealed two subgroups characterised by a significant association between cluster membership and presence of COPD (p=0.024). Ontology analysis of genes containing differentially methylated CpGs (false discovery rate, FDR-adjusted p<0.05) revealed that immune genes were strongly enriched in COPD tumours, but not in non-COPD tumours. This COPD-specific immune signature was attributable to methylation changes in immune genes expressed either by tumour cells or tumour-infiltrating immune cells. No such differences were observed in adjacent tissue. Transcriptome profiling similarly revealed that genes involved in the immune response were differentially expressed in COPD tumours (FDR-adjusted p<0.05), an observation that was independently replicated using TCGA data. Immunohistochemistry validated these findings, revealing fewer CD4-positive T lymphocytes in tumours derived from patients with COPD.
Conclusions Lung tumours of patients with COPD differ from those of patients without COPD, with differentially methylated and expressed genes being mainly involved in the immune response.
- COPD ÀÜ Mechanisms
- Innate Immunity
- Lung Cancer
Statistics from Altmetric.com
Key messages
What is the key question?
Although COPD independently increases the risk for non-small cell lung cancer (NSCLC) up to fivefold, it has not been established how COPD influences NSCLC development and tumour progression.
What is the bottom line?
Lung tumours in patients with COPD are, in contrast to those from patients without COPD, characterised by an immune-related signature as revealed by unbiased DNA methylation profiling, and confirmed by gene expression profiling and immunohistochemistry.
Why read on?
The identified immune-related signature in COPD-associated NSCLC may contribute to the optimisation of novel NSCLC treatment strategies by assisting in treatment stratification and prediction of treatment response.
Introduction
Lung cancer is the leading cause of cancer-related death worldwide with an alarmingly low 5-year survival rate of <15%. Therapies targeting specific genetic lesions in the tumour are increasingly being used to treat lung cancer, highlighting the need to accurately classify lung tumours based on their molecular-genetic composition.1 Similar to somatic mutations, epigenetic modifications may provide a substrate for clonal selection during malignant transformation. As such, genome-wide methylation profiling studies can identify signatures that contribute to the molecular subclassification of various cancers.2 For instance, methylation profiling classifies breast tumours into its four established expression subtypes and has already identified an additional subtype associated with T lymphocyte infiltration and favourable outcome.3
Over 50% of patients with lung cancer also suffer from COPD due to shared risk factors, such as age and cigarette smoking.4 However, COPD also independently increases the risk for lung cancer up to fivefold.5 Although genetic association studies revealed susceptibility variants overlapping between both diseases,6 ,7 the precise mechanisms underlying this link are incompletely understood.8 Here, we hypothesise that the inflammatory microenvironment in COPD lungs, which results from a maladaptive response of the immune system to inhaled noxious particles, may influence lung cancer development through its impact on the epigenome. Indeed, among various biological processes, changes in immune response-related genes were recently observed in peripheral blood leucocytes and bronchial small airway epithelial cells of cancer-free smokers with COPD.9 Likewise, age-related CpG island hypermethylation in non-malignant human tissues and gene-specific hypomethylation in blood of active smokers have been described.10 Surprisingly, although lung cancer-specific methylation profiles have previously been generated, the potential contributions of COPD, age and smoking history to these signatures have not been assessed.
Methods
Additional details can be found in the online supplementary material.
Study participants
We prospectively recruited untreated patients with non-small cell lung cancer (NSCLC) scheduled for curative-intent surgery and a smoking history of ≥15 pack-years at the Leuven University Hospital (Belgium) between March 2010 and August 2011. Prior to surgery, all 49 eligible participants performed spirometry according to the American Thoracic Society and European Respiratory Society guidelines.11 COPD was defined as postbronchodilator FEV1/FVC<0.70 and staged according Global Initiative for Chronic Obstructive Lung Disease 2007 guidelines.12 Emphysema was semiquantitatively assessed by a radiologist, as described previously.6 ,7 Other respiratory diseases affecting pulmonary function were excluded. Previous treatments for COPD were retrieved from electronic medical files. The study protocol was approved by the local ethical committee. All participants provided written informed consent.
DNA isolation and methylation profiling
DNA was extracted from fresh-frozen tumour tissue, in which ≥70% of the epithelial cells were malignant based on histological examination, and adjacent lung tissue from the same resection specimen at maximal distance (>5 cm) from the tumour. Bisulfite-converted DNA was hybridised on 450 K BeadChips, using the Infinium HD methylation protocol. After controlling for batch effect and normalising raw intensity data, the methylation level of each interrogated CpG, scored as a β value between 0 and 1, was calculated according to the fluorescent intensity ratio.
RNA extraction and transcriptome sequencing
Total RNA was extracted from all tumour samples undergoing methylation profiling. RNA sequencing was performed using Illumina HiSeq2000. In addition, RNA sequencing data of tumour and adjacent lung tissue samples of 178 patients with NSCLC were downloaded from the The Cancer Genome Atlas (TCGA) portal (https://tcga-data.nci.nih.gov/tcga/).
Histological analyses
An experienced pathologist blinded to the clinical data semiquantitatively scored H&E-stained tumour sections for immune cell infiltrates, as no-to-mild or moderate-to-severe infiltration present. Consecutive tumour sections were stained with antibodies to human T cells (CD3, CD4, CD8), B cells (CD20), macrophages (CD68), neutrophils (myeloperoxidase (MPO), CD66b) and natural killer cells (neural cell adhesion molecule, (NCAM)). Stained sections were screened at low power (×100) to identify five areas with the highest density of positive cells (hot spots). Next, at each hot spot, an image was taken at ×400 magnification (high power field, HPF) and the absolute number of positive cells per hot spot surface was counted manually to obtain the average density of positive cells per 5 HPFs (positive cells/mm2).
Statistical analyses
Methylation analyses were performed on normalised β values. Top-down unsupervised hierarchical clustering was performed using a recursively partitioned mixture model (RPMM).13 Multivariate logistic regression models with forward stepwise selection assessed age, histological subtype, pack-years smoked, years-quit smoking, COPD status, emphysema, COPD treatment and batch as independent predictors. Differential methylation was assessed between tumour and adjacent lung tissue in the entire study population, in COPD and non-COPD subgroups. We used a paired linear model for differential methylation assessment (Limma). A CpG site was differentially methylated when exhibiting a false discovery rate (FDR)-adjusted p value <0.05 and a difference in β value between tumour and adjacent lung tissue (Δβ value) >0.075.14 Differential expression analysis was performed using DESeq following normalisation with EDASeq. A twofold or greater change in expression and FDR <0.05 were selected to designate significantly upregulated or downregulated genes. Gene ontology (GO) enrichment analysis was performed using GOrilla. Only GO-terms with a FDR <0.05 were considered as significantly enriched. The association of COPD with the number of tumour-infiltrating immune cells was assessed by linear regression adjusted for covariates. Statistical analyses were performed using SPSS software (V.19.0) and R (V.2.14.1).
Results
Novel NSCLC subtypes based on DNA methylation profiling
We included 49 patients with NSCLC that underwent curative-intent surgery. Tissue pairs from three patients were excluded due to low signal intensities for some methylation probes, as described in the online supplementary material. Clinical characteristics of the remaining 46 patients are shown in table 1.
Clinical characteristics of the study population and of DNA methylation-based tumour clusters
Pairwise comparison of methylation values between tumour and adjacent lung tissue (Δβ values) revealed tumour-associated methylation changes similar to those previously described (see online supplementary results and figure S1).15 For instance, tumour-associated hypermethylation occurred three times more frequently in gene promoters targeted by Polycomb group proteins than in other promoters (ie, 26.9% vs 8.8%, p<0.001).16
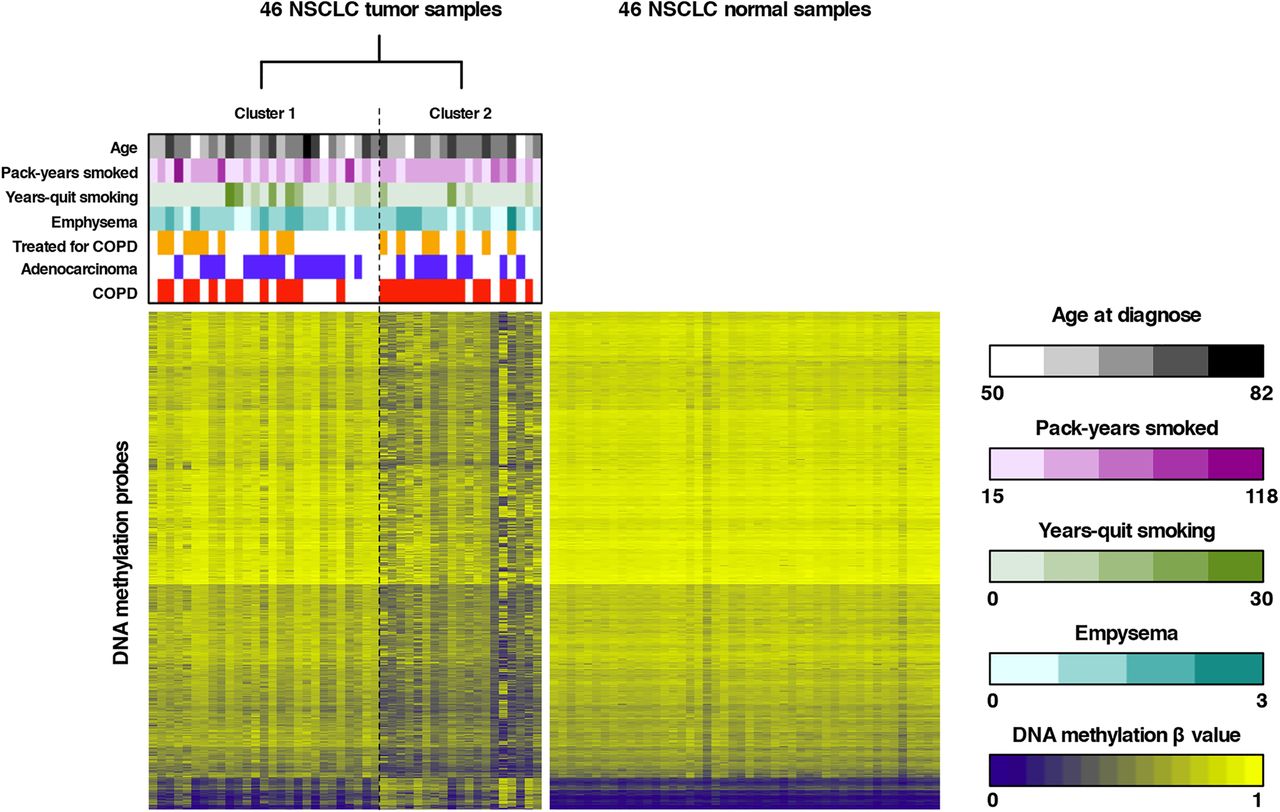
To subsequently identify novel NSCLC subtypes, unsupervised RPMM-based clustering of all tumour β values that passed quality control was performed, revealing two major clusters (see figure 1 and online supplementary results and table S1). Forward-logistic regression established an independent association between tumour cluster membership and COPD status (p=0.024), but not with other covariates in the regression model (age, histological subtype, pack-years smoked, years-quit smoking, emphysema, COPD treatment and batch). More specifically, we found that one cluster was significantly enriched in patients with COPD (15/19 or 78.9% vs 12/27 or 44.4%; table 1). Similarly, clustering of β values in adjacent lung tissue also revealed two clusters, one of which was enriched in patients with COPD (78.6% vs 50.0%). COPD status did, however, not significantly influence cluster membership, as assessed by forward-logistic regression (online supplementary table S2). There was no association between other clinical variables and cluster membership. Additionally, no overlap in cluster membership between the tumour-based and adjacent tissue-based clusters was noted.
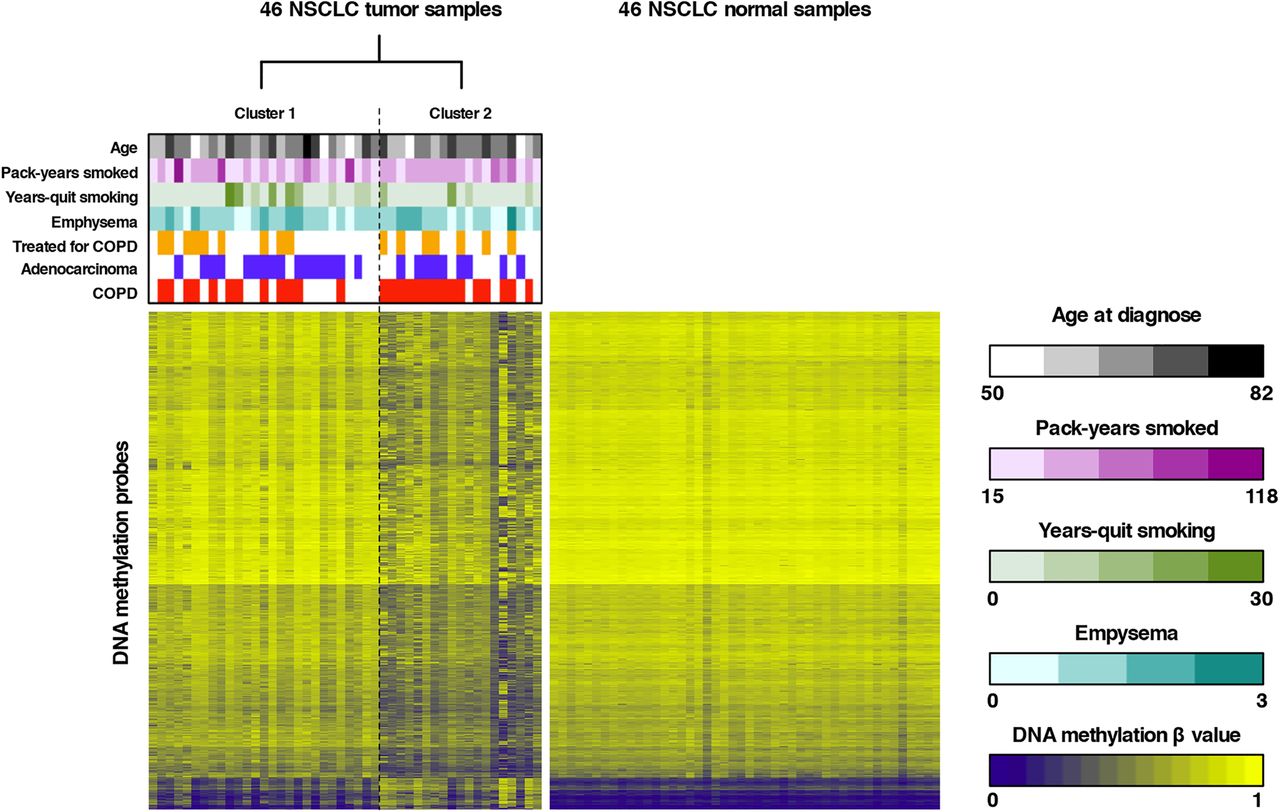
Unsupervised clustering of tumour β values and heatmap representation of DNA methylation data. Left panel: recursively partitioned mixture model (RPMM)-based classification of 46 non-small cell lung cancer (NSCLC) tumour samples (columns) and heatmap representation of the 13 965 most variable β values (rows) between the two clusters (false discovery rate, FDR <0.01 and Δβ value between both clusters >0.15). β values are represented by a colour scale ranging from dark blue (low DNA methylation) to bright yellow (high DNA methylation). Clustering revealed two tumour clusters (n=27 and n=19 for clusters 1 and 2, respectively), which differed according to COPD status, as indicated above the heatmap. Samples from patients with COPD are indicated in red above the heatmap, adenocarcinoma samples are indicated in blue and patients receiving COPD treatment are indicated in orange. Continuous variables (age, pack-years smoked and years-quit smoking) and the degree of emphysema are represented by colour scales (respectively, black, purple, green and turquoise) ranging from light (low number) to dark (high number). Right panel: For comparison, a heatmap representation of the 13 965 β values identified by clustering of tumour β values (left panel) in the 46 adjacent non-malignant lung tissues is shown. All adjacent lung tissue samples have comparable methylation values for the selected tumour-associated CpGs.
Differential methylation according to COPD status
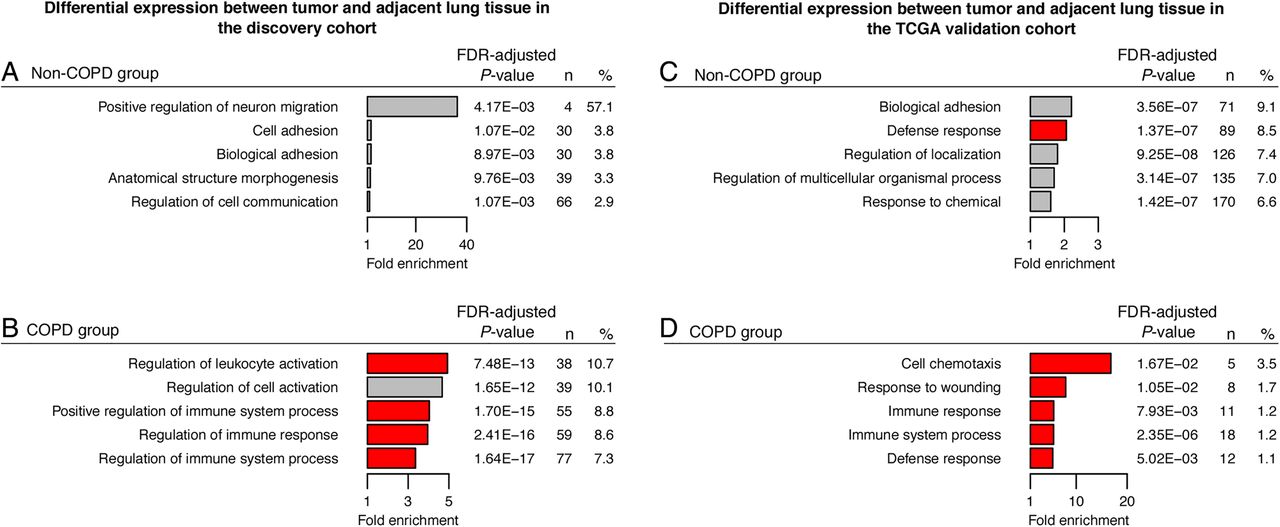
Since COPD status differed significantly between cluster subgroups, we stratified our study population by COPD status. Clinical characteristics of COPD and non-COPD subgroups are presented in online supplementary table S3. First, to identify COPD-associated methylation events, we compared for each CpG the β values between tumour versus adjacent lung tissue in the COPD and non-COPD subgroups separately. Subsequently, we selected for both subgroups those CpGs that displayed a significant difference in Δβ value (Δβ >0.075 and FDR <0.05 using a paired linear model; online supplementary table S1) and performed a GO enrichment analysis. In the COPD subgroup, genes with CpGs that were hypermethylated in tumour versus adjacent tissue were enriched for GO terms involved in immune response activation, whereas no enrichment was observed in the non-COPD subgroup (figure 2A, C). On the other hand, genes with hypomethylated CpGs were not enriched for GO terms in the COPD subgroup, whereas a diverse set of GO processes was identified in the non-COPD subgroup (figure 2B, D).

Gene ontology (GO) terms enriched in genes differentially methylated in patients with non-small cell lung cancer (NSCLC) with and without COPD. GO enrichment analysis was performed on genes containing significantly differentially methylated β values in tumour versus adjacent tissue in the COPD subgroup (left panel) or in the non-COPD subgroup (right panel). Left panel: For the COPD subgroup: (A) the 5 most significantly enriched GO terms among the 546 hypermethylated genes are presented; (B) no GO enrichment was identified among the 2154 hypomethylated genes. Right panel: For the non-COPD subgroup: (C) no GO enrichment was identified among the 1835 hypermethylated genes; (D) the 5 most significantly enriched GO terms among the 1169 hypomethylated genes are presented. Horizontal bars represent the fold enrichment (x axis) for the significantly enriched GO terms, and bars for GO terms associated with immunological processes are red. The false discovery rate (FDR) values for enrichment, the number of differentially methylated genes associated with a specific GO term (n), and the ratio of n and the total number of genes associated with a specific GO term (%) are provided at the right of the associated bars.
To assess whether the enrichment for immune response genes in patients with COPD originated from tumour or adjacent lung tissue, we directly compared tumour β values between patients with and without COPD. GO analysis of genes relatively hypermethylated in COPD tumours (p<0.05; online supplementary table S1) confirmed that these genes were significantly enriched for immune genes. When comparing adjacent lung tissue between both subgroups, no enrichment for GO terms was found, which is in accordance with the less-pronounced differences in methylation-based clusters in adjacent lung tissue. Hence, hypermethylation of immune response genes in patients with COPD specifically affects the tumour tissue.
In a next step, we assessed whether the immune signature in COPD tumour tissue was attributable to a difference in the amount of immune cells infiltrating the tumour or to differential methylation of immune-related genes expressed by tumour cells. We found evidence for both mechanisms. In particular, using the Immunological Genome Project database,17 we found that genes specifically expressed by immune cells, such as CD4, which is expressed by mature T helper (TH) cells, exhibited promoter hypermethylation in COPD tumours. Since promoters of expressed genes are usually not methylated, their high methylation status in COPD tumours suggests that there is reduced infiltration of immune cells in these tumours. For other immune genes, we found published evidence that they are predominantly expressed in tumour cells and that changes in their expression levels influence the antitumour immune response. These genes included CCL5, encoding a chemoattractant; TNFRSF21, encoding a member of the tumour necrosis factor (TNF) receptor superfamily; and SUSD2, influencing tumour infiltration by CD4-positive lymphocytes. Hypermethylation of the latter genes in patients with COPD is therefore likely to occur in tumour cells.
Methylation-driven gene expression changes
Next, to investigate the impact of tumour-associated DNA hypermethylation on gene expression, we sequenced mRNA of 39 tumour-normal pairs (other pairs failed RNA quality control assessment; see online supplementary results). On average, 36.0 million sequence reads were generated per sample and 69.4% of coding genes were expressed.
Given the observed differences in methylation between tumours in patients with and without COPD, we stratified expression data for COPD status (21 and 18 patients with and without COPD, respectively; online supplementary table S4). Moreover, since immune response genes were hypermethylated in COPD tumours and DNA hypermethylation of gene promoters results in silencing of the associated gene, we focused on genes downregulated in tumour versus adjacent tissue (FDR<0.05; online supplementary table S1). GO-analysis of genes characterised by reduced expression in the tumour revealed a strong enrichment of immune genes in the COPD subgroup only, confirming that the observed hypermethylation events in COPD tumours were relevant at the expression level (figure 3A, B). Notably, also after correcting for COPD treatment, GO analysis revealed the same enrichment for immune genes. In adjacent tissue, gene expression changes did not show enrichment for immune-related GO terms in patients with versus without COPD, although a significant enrichment in immune-related GO terms among genes significantly downregulated (p<0.05) was noted in adjacent tissue from treated versus non-treated patients with COPD (online supplementary figure S2), as reported previously.18 However, since a less stringent p value threshold was used to select downregulated genes in this analysis, this effect is clearly less pronounced than the immune-related signature in COPD tumours.

Gene ontology (GO) terms enriched in genes differentially expressed in patients with non-small cell lung cancer (NSCLC) with and without COPD. GO enrichment analysis was performed on genes significantly downregulated in tumour versus adjacent lung tissue from patients with NSCLC with COPD (n=445 genes, A) and without COPD (n=295 genes, B) in our RNA-seq discovery cohort (left panel); and on genes significantly downregulated in tumour versus adjacent lung tissue from patients with NSCLC with COPD (n=34 genes, C) and without COPD (n=698 genes, D) in the RNA-seq The Cancer Genome Atlas (TCGA) validation cohort (right panel). The 5 most significantly enriched GO terms for each subgroup are presented. Horizontal bars represent the fold enrichment (x axis) for the significantly enriched GO terms, and bars for GO terms associated with immunological processes are red. The false discovery rate (FDR) values for enrichment, the number of differentially expressed genes associated with a specific GO term (n), and the ratio of n and the total number of genes associated with a specific GO term (%) are provided at the right of the associated bars.
Next, we integrated DNA methylation and RNA sequencing data obtained from patients with COPD (see online supplementary figure S3). Since GO terms were not enriched in hypomethylated genes when comparing COPD tumour versus adjacent lung tissue, we focused on hypermethylated genes. Of the 1985 genes with promoter hypermethylation in tumour versus adjacent tissue (FDR<0.05), we identified 311 genes whose expression was more than twofold downregulated in tumour versus adjacent tissue of patients with COPD (FDR<0.05; online supplementary table S1). These contained 76 immune-related genes (figure 4A, B).
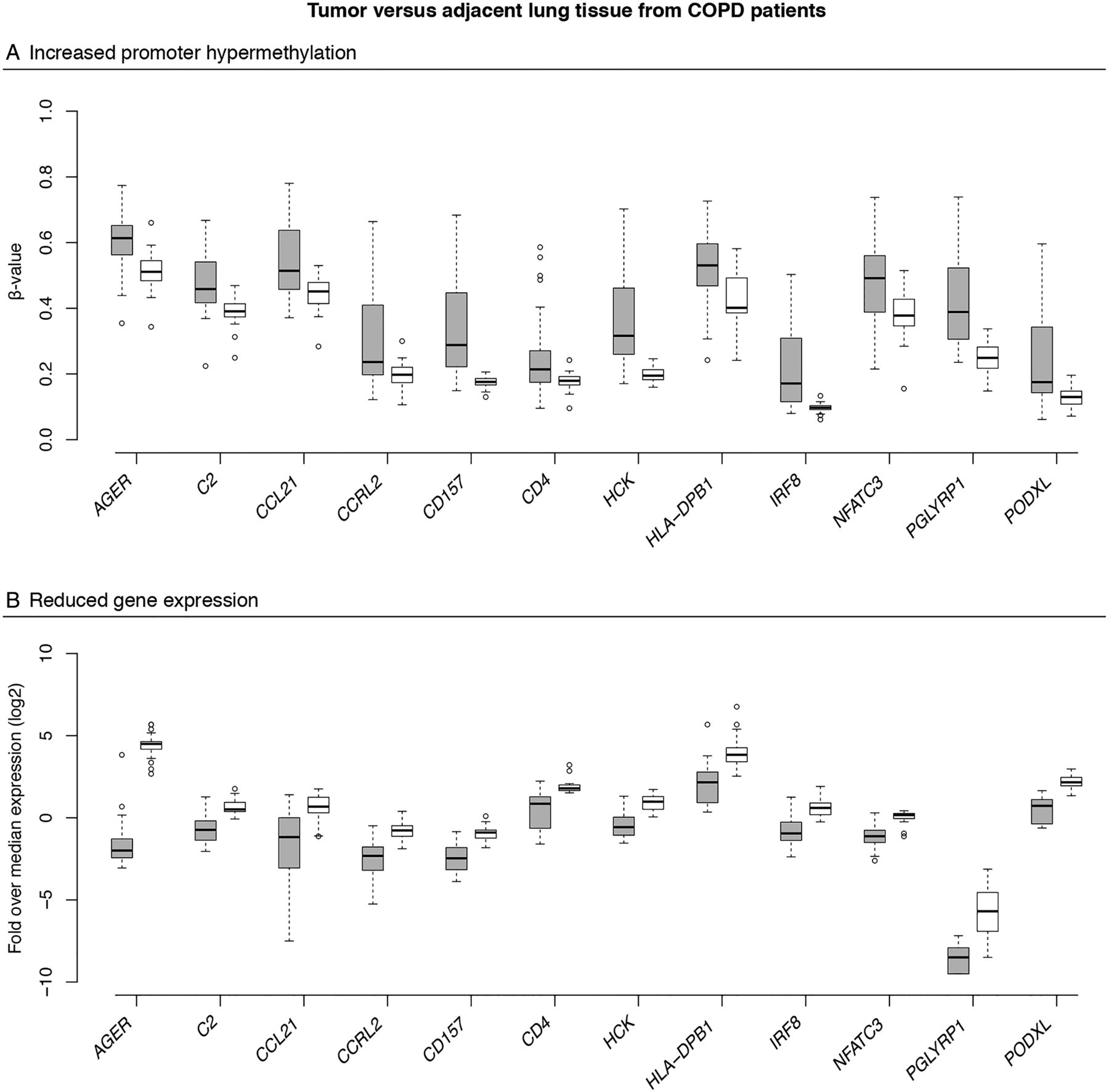
Integration of DNA methylation and gene expression changes. Box plots showing changes in promoter DNA methylation (A) and gene expression (B) between tumour tissue (grey boxes) and adjacent lung tissue (white boxes) from patients with non-small cell lung cancer (NSCLC) with COPD for 12 immune genes found to be promoter hypermethylated and downregulated, including CD157 involved in migration of neutrophils to the inflammation site and CCRL2 involved in lung dendritic cell trafficking and priming of a T helper cell response. The y axis represents the mean promoter β value (A) and the gene expression as the log2-transformed fold change over median gene expression level for each sample (B) for the selected genes. The box represents the IQRs. The line across the box indicates the median. The whiskers extend from the box to the 90th and 10th centiles, excluding outliers. Outliers (open circles) are defined as cases outside the 90th and 10th centiles. For all selected genes, methylation and gene expression differed significantly between tumour and adjacent lung tissue from patients with COPD (false discovery rate, FDR-adjusted p values <0.05).
Validation of an immune-specific signature in patients with lung cancer and COPD
As an independent validation of the COPD-specific expression signature, we used TCGA expression data of 178 patients with NSCLC for whom spirometry data were available.19 After stratification of this cohort for COPD status (46 patients with and 132 patients without COPD; online supplementary table S5), GO analysis of genes downregulated in tumour versus adjacent tissue (FDR <0.05; online supplementary table S1) confirmed the enrichment of immune genes in the COPD subgroup, thus independently validating the presence of an immune-specific signal in COPD tumours (figure 3C, D).
Finally, we also assessed which immune genes were downregulated in tumour versus adjacent tissue from patients with COPD in our discovery and validation cohorts (FDR <0.05 in both cohorts). Remarkably, of the 535 genes that were significantly downregulated in COPD tumours in both cohorts, 161 genes were associated with an immune-related GO term (online supplementary table S1). Interestingly, of the 311 genes that were hypermethylated and downregulated in our discovery cohort, 125 genes were also downregulated in tumour versus adjacent tissue in the validation cohort (FDR <0.05). Genes were involved in the innate defence response (AGER), lung dendritic cell trafficking (CCRL2), lymphocyte migration (S1PR1), etc (figure 5).

Expression of selected immune markers downregulated in the RNA-seq discovery and validation cohort. Box plots showing gene expression levels of 12 immune genes with a significantly lower expression level in tumour tissue from patients with COPD (grey boxes) versus tumour tissue from patients without COPD (white boxes), in the discovery and The Cancer Genome Atlas (TCGA) validation cohorts (false discovery rate (FDR)<0.05 for both). The y axis represents the gene expression as the log2-transformed fold change over median gene expression level for each sample. The box represents the IQRs.
Histopathology of immune cell infiltrates in COPD versus non-COPD tumours
We also quantified infiltrating immune cells using histopathology. First, an experienced pathologist estimated the degree of immune cell infiltration on H&E-stained sections from all 49 tumours selected for methylation profiling. This revealed significantly less immune cell infiltration in patients with COPD (p=0.036; figure 6A–C and online supplementary figure S4). Multivariable-adjusted logistic regression confirmed that this effect was independent from covariates (p=0.022).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fewer tumour-infiltrating immune cells in patients with COPD versus patients without COPD. (Top panel) Representative microscopic pictures showing different degrees of immune cell infiltration in H&E stained tumour sections. (A) Tumour section with mainly fibrosis (indicated by the white arrow) and a low density of immune cell infiltrates between tumour cell nests (some of which are marked by a white asterisk). This section is representative for tumour sections with no-to-mild immune cell infiltration. (B) Tumour section with pronounced immune cell infiltration (indicated by the white arrow) between the tumour cell nests (some of which are marked by a white asterisk). This section is representative for tumour sections with moderate-to-severe immune cell infiltration. (C) Stacked bar plot showing the proportion of tumours showing no-to-mild versus moderate-to-severe (coloured in grey) immune cell infiltration between patients with COPD and patients without COPD. A significantly higher proportion of COPD tumours showed no-to-mild immune cell infiltration. (Middle panel) Representative pictures of tumour sections from patients with COPD (D) and patients without COPD (E) stained with an antibody directed against CD3. Positive cells are coloured brown. (F) Dot chart showing the density of tumour-infiltrating CD3-positive cells in patients with COPD versus patients without COPD. The y axis represents the average density of positive cells per 5 high power fields (HPFs). The horizontal black bar represents the mean. (Lower panel). Representative pictures of tumour sections from patients with COPD (G) and patients without COPD (H) stained with an antibody directed against CD4. Positive cells are coloured brown. (I) Dot chart showing the density of tumour-infiltrating CD4-positive cells in patients with COPD versus patients without COPD. The y axis represents the average density of positive cells per 5 HPFs. The horizontal black bar represents the mean. Asterisks (*) indicate p<0.05. Original magnifications: ×50 (A,B) and ×400 (D,E,G,H). Scale bar, 200 µm (upper panel) and 50 µm (middle and lower panel).
Second, we stained consecutive tumour sections for established T cell (CD3, CD4, CD8), B cell (CD20), neutrophil (MPO, CD66b), macrophage (CD68) and natural killer cell (NCAM) markers. Multivariable-adjusted linear regression revealed a significant association between COPD status and the average density of CD3-positive T cells (p=0.001). In particular, tumours from patients with COPD contained fewer CD3-positive T cells (figure 6D–F). There was a similar correlation between COPD status and the density of CD4-positive cells (p=0.012; figure 6G–I). CD4 is commonly used as a TH cell marker. However, it can also be expressed by other immune cells, such as macrophages. The fact that we observed reduced infiltration for CD3-positive and CD4-positive cells suggests that infiltration of CD4-positive TH cells in COPD tumours was reduced. This was confirmed microscopically based on typical TH cell morphology. Furthermore, there were no significant differences observed for the CD68 macrophage-specific marker (see online supplementary figure S5).
Finally, to elucidate whether specific TH cell subtypes were affected, we reanalysed gene expression data from the discovery cohort. In particular, by assessing TH cell subtypes (TH1, TH2, TH9, TH17, TH22, regulatory (TReg) and follicular (TFH) T cells) using a published marker set20 (see online supplementary table S6), we observed that global expression of TH cell markers in COPD tumours was indeed reduced (p=0.018). Particularly, at the individual TH cell subtype level, expression of TReg markers was significantly reduced in COPD tumours (p=0.046, online supplementary figure S6).
Discussion
We studied 49 pairs of surgically resected tumours and adjacent tissues using genome-wide methylation profiling. Unsupervised clustering of tumour data identified two subgroups that represented different populations in terms of COPD status. Although other clinical covariates are also likely to affect the methylation profile of the tumour, our data suggest that the effect of COPD is stronger than that of age, smoking behaviour, emphysema and histopathological subtype. This is surprising, because age and smoking are major risk factors for NSCLC, and because histopathology reflects the difference in cell type affected by the carcinogenic process, whereby cell type identity is a well-known determinant of DNA methylation. In the adjacent tissue, no such difference was observed in patients with COPD versus patients without COPD, thereby confirming previous DNA methylation and gene expression studies.21–26 Only when stratifying for COPD treatment, a weak enrichment in immune-related GO terms was observed for genes downregulated in the adjacent tissue of the treated subgroup of patients with COPD, as reported by others.18 COPD thus seems to more strongly affect DNA methylation of tumour than of normal tissue.
Stratification for COPD status revealed that epigenetic events in COPD-associated NSCLC were enriched for immune-related genes. This was attributable to a relative hypermethylation of immune genes either predominantly expressed by tumour-infiltrating immune cells or by tumour cells. Promoters of genes expressed by immune cells are mostly unmethylated, but may become hypermethylated (and thus not expressed) in tumour cells. A higher methylation status of such genes thus suggests that there is reduced infiltration of immune cells in the tumour. This was indeed confirmed by immunohistopathology. Combined with our observation that also immune genes predominantly expressed by tumour cells were hypermethylated, these findings suggest that COPD tumours are characterised by a less pronounced immune response.
Since COPD lungs are characterised by increased immune cell infiltration and excessive chronic inflammation,27 these findings may look surprising. However, often the immune system of patients with COPD is largely dysfunctional. Their macrophages, for instance, exhibit deficient phagocytic functions towards apoptotic epithelial cells and bacteria, thereby sustaining chronic inflammation and predisposing to infections that are characteristic of severe COPD.27–30 The immune system in patients with COPD may thus not be capable of mounting an effective immune response against the tumour. As a result, in COPD tumours, the infiltration of TH cells will be less pronounced, which will further weaken antitumour immunity, as TH cells are essential for inducing a strong immune response. Accordingly, the more pronounced downregulation of markers for TReg cells, which in principle have a tumour-associated immunosuppressive effect, supports the hypothesis that COPD tumours are less dependent on immunosuppression.
Overall, we thus identified a NSCLC subtype characterised by reduced immune cell infiltration and associated with COPD status. Interestingly, cancer subtypes characterised by differential expression of immune response genes have previously been established as predictors of treatment response and survival in other cancers.31–33 Future studies assessing novel NSCLC treatment strategies should thus take COPD status or reduced immune cell infiltration as a potential predictive biomarker into account. Especially when testing cancer immunotherapies, COPD status might have to be considered. Very recently, US Food and Drug Administration (FDA) approved the anti-PD-1 therapy, nivolumab, as the first immunotherapy for metastatic NSCLC.34 It would obviously be very interesting to assess whether patients with NSCLC respond differently to this novel therapy depending on their COPD status.
Although methylation profiling studies have previously been conducted for NSCLC tumours, our study is unique in several respects. First of all, our patient population was extensively phenotyped and for the first time included COPD status as a covariate (see online supplementary table S7). Second, adjacent non-malignant lung tissue was collected for each tumour sample, allowing us to correct for interindividual epigenetic variation due to aging and smoking. Indeed, immune-related GO terms were more enriched when analysing Δβ values than tumour β values. A potential limitation is that there were no methylation data publicly available to replicate our clustering results. Additionally, as it is virtually impossible to collect lung tissue from cancer-free patients without COPD (because these patients do not undergo surgery), we were forced to compare COPD versus non-COPD adjacent lung tissue from patients with NSCLC undergoing surgery. Effects of the tumour on the adjacent resection specimen can thus not formally be excluded. Finally, systemic corticosteroid use has been shown to influence DNA methylation in patients with COPD.35 However, although other COPD treatments also affected gene expression, we were not able to explore this specifically for systemic corticosteroids given the limited number of patients treated (n=4).
Overall, our study describes the presence of a specific tumour microenvironment in COPD-associated NSCLC, which is characterised by reduced immune cell infiltration, thereby providing novel insights into how lung tumours of patients with COPD differ from those of patients without COPD.
Acknowledgments
The authors thank Thomas Van Brussel, Gilian Peuteman, Stephan Vinckier, Kristien De Bent, Maarten Spruyt, Erica Balligand, An Lehouck and Beatrijs Anrijs for their excellent assistance during data collection and for technical support. The authors also thank the lung function technicians from the UZ Leuven for their dedication and efforts.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
Footnotes
EW, MM and DL contributed equally to this paper.
Contributors Conception and design: WJ, JV and DL; Experimental work and data analysis: EW, MM, HD, NH, HZ, JC, DS, XS and PDL; Data interpretation and manuscript writing: EW, BT, WJ, JV, MD, AL, MM and DL. All authors read and approved the final version of the manuscript.
Funding EW, MM, HZ and BT are supported by the Fund for Scientific Research Flanders (FWO-F). MM is supported by SymBioSys. WJ is clinical researcher of FWO-F. This work was supported by a European Research Council (ERC) consolidator grant RCN:191995 (DL), by a FWO-F research project G065615N and by Vlaamse Liga tegen Kanker (VLK).
Competing interests None declared.
Ethics approval Ethical Comity University Hospital Gasthuisberg.
Provenance and peer review Not commissioned; externally peer reviewed.