Article Text

Abstract
Background The phenotypic spectrum of cystic fibrosis (CF) has expanded to include patients affected by single-organ diseases. Extensive genotyping and nasal potential difference (NPD) testing have been proposed to assist in the diagnosis of CF when sweat testing is inconclusive. However, the diagnostic yield of extensive genotyping and NPD and the concordance between NPD and the sweat test have not been carefully evaluated.
Methods We evaluated the diagnostic outcomes of genotyping (with 122 mutations included as disease causing), sweat testing and NPD in a prospectively ascertained cohort of undiagnosed patients who presented with chronic sino-pulmonary disease (RESP), chronic/recurrent pancreatitis (PANC) or obstructive azoospermia (AZOOSP).
Results 202 patients (68 RESP, 42 PANC and 92 AZOOSP) were evaluated; 17.3%, 22.8% and 59.9% had abnormal, borderline and normal sweat chloride results, respectively. Only 17 (8.4%) patients were diagnosable as having CF by genotyping. Compared to sweat testing, NPD identified more patients as having CF (33.2%) with fewer borderline results (18.8%). The level of agreement according to kappa statistics (and the observed percentage of agreement) between sweat chloride and NPD in RESP, PANC and AZOOSP subjects was ‘moderate’ (65% observed agreement), ‘poor’ (33% observed agreement) and ‘fair’ (28% observed agreement), respectively. The degree of agreement only improved marginally when subjects with borderline sweat chloride results were excluded from the analysis.
Conclusions The diagnosis of CF or its exclusion is not always straightforward and may remain elusive even with comprehensive evaluation, particularly among individuals who present at an older age with single-organ manifestations suggestive of CF.
- Cystic Fibrosis
- Bronchiectasis
- Paediatric Lung Disaese
Statistics from Altmetric.com
Key messages
What is the key question?
-
Does extensive genotyping and NPD testing help clarify and improve the diagnostic yield of CF over sweat testing alone in patients with single organ manifestations of CF?
What is the bottom line?
-
The diagnosis of CF may remain inconclusive despite comprehensive evaluation by sweat testing, extensive genotyping and NPD testing.
Why read on?
-
Extensive CFTR genotyping was the least sensitive diagnostic test even when 122 additional mutations defined by CFTR2 as disease causing were included. While NPD testing identified more patients diagnosable with CF and fewer patients with borderline results in comparison with the classic sweat test, there was considerable test discordance (sweat test normal but NPD abnormal and vice versa).
In early childhood the diagnosis of cystic fibrosis (CF) is usually straightforward in the presence of a CF phenotype and sweat chloride concentration of 60 mmol/L or greater. However, since the discovery of the CF transmembrane conductance regulator (CFTR) gene,1 an expanded spectrum of conditions has been associated with CFTR mutations, particularly in older children and adults who present with single-organ manifestations such as sino-pulmonary diseases, pancreatitis or obstructive azoospermia.2–9 The majority of these individuals are pancreatic sufficient (PS).
The sweat test remains the principal test for the diagnosis of CF.10 ,11 As the reference range for sweat chloride was originally defined by evaluating children presenting with classic multi-organ symptoms,12–15 it is not surprising this test satisfactorily discriminates the majority of pancreatic insufficient (PI) CF patients from unaffected individuals. However, sweat testing may be inconclusive in a subset of patients, especially those who present at an older age with single-organ manifestations of CF.4 ,5 ,8–10
Several strategies such as extensive genotyping and nasal potential difference (NPD) testing have been proposed to reduce diagnostic dilemmas.10 ,11 ,16 Until recently, genotyping has been limited by the fact that only 23 mutations were designated as CF causing.10 In fact, most patients presenting at an older age typically carry mutations that were not listed as disease causing. The clinical and functional translation of CFTR (CFTR2) project recently expanded the list to 122 CF-causing mutations.17 CFTR2 also described mutations of ‘varying clinical consequence’, because they had been identified in individuals with and without CF. The diagnostic yield of this greatly expanded number of mutations has not been evaluated. NPD testing, which measures the bioelectric properties of the nasal epithelium, was first established for research purposes. It has also been recommended as a diagnostic tool in individuals with an uncertain diagnosis of CF. Nevertheless, NPD is limited in clinical practice by lack of availability and diagnostic reference values, and may still lead to inconclusive outcomes. According to American and European consensus recommendations, NPD is only recommended as the next test when patients have ‘borderline’ sweat chloride concentrations.10 ,11 ,16 NPD testing is thus not recommended when the sweat test is normal (<40 mmol/L) or abnormal (>60 mmol/L). This assumes that ‘clear-cut’ normal or abnormal sweat test results would concur with the NPD result. However, the concordance between the two tests has never been assessed.
Due to the aforementioned issues, we evaluated and compared the diagnostic outcomes of sweat testing, genotyping and NPD in a prospectively ascertained cohort of undiagnosed patients who presented with single-organ manifestations suggestive of CF. Reference cohorts of non-CF and established CF subjects were also evaluated for comparison. In particular, we aimed to investigate the diagnostic yield from genotyping using CFTR2's expanded list of CF-causing mutations and to assess the contribution and concordance between sweat chloride and NPD results.
Methods
Subjects
Two cohorts were prospectively and consecutively enrolled from the Toronto CF clinics from 1994 to 2008: undiagnosed patients (>5 years) with a single-organ manifestation of CF, and reference cohorts consisting of non-CF (healthy controls and obligate heterozygotes) and CF (CFPS and CFPI) subjects.18 All subjects were evaluated by sweat test, CFTR genotyping and NPD (figure 1); subjects unable to complete all three tests were excluded from analyses.

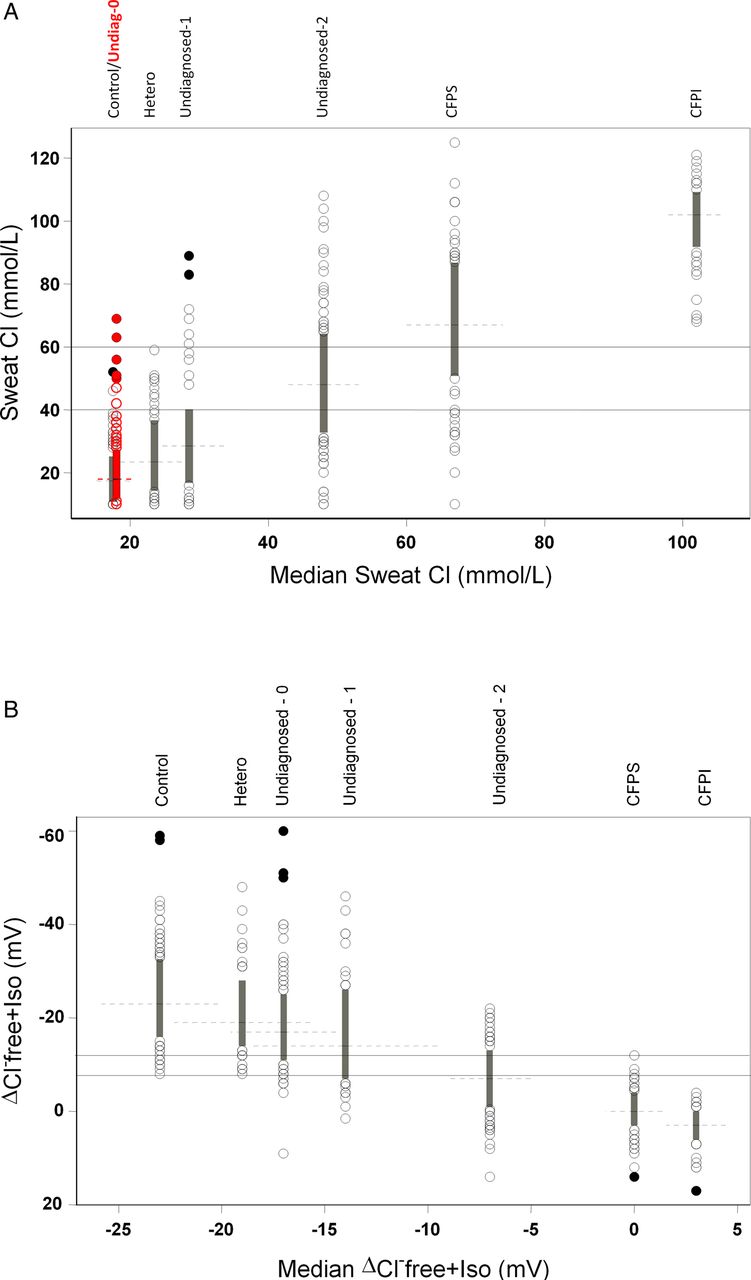
The range of each measurement and the relationship of each group with one another are demonstrated by arranging box plots for each group along the x axis according to the value of the median for: (A) sweat chloride and (B) ΔCl-free+Iso. Each box plot represents values within the 25th to 75th percentiles (IQR). Values outside the 25th and 75th percentile are represented by circles. Outliers, values that are more than 1.5 IQR above and below the 75th and 25th percentiles, respectively, are represented by solid circles. Horizontal dashed lines depict the medians of each group. Due to the near-superimposition between control and undiagnosed-0 box plots in Figure 1A, Undiagnosed-0 subjects are plotted in red (see online material). CF, cystic fibrosis; CFPI, pancreatic insufficient; CFPS, pancreatic sufficient; Undiagnosed-0, subjects who presented with a single organ CF phenotype and not identified with any CFTR mutation; Undiagnosed-1=subjects who presented with a single organ CF phenotype and one mutation; Undiagnosed-2, subjects who presented with a single organ CF phenotype and two mutations.
Undiagnosed patients with suspected CF included those with: idiopathic chronic sino-pulmonary disease (RESP); idiopathic recurrent acute/chronic pancreatitis (PANC); or infertile men with obstructive azoospermia (AZOOSP) (see supplementary material, available online only). Healthy controls consisted of volunteers without a past or family history of CF, pancreatic disease and male infertility. No healthy controls were excluded on the basis of age, sex or race. Heterozygotes consisted of parents/siblings of CF patients. Reference subjects with a previous diagnosis of CF based on the US Cystic Fibrosis Foundation consensus criteria had ion channel measurements derived prospectively, irrespective of previously performed test(s).10 These subjects were included in a report comparing the American and European diagnostic guidelines for CF.18
Ion channel measurements
Sweat test and NPD were performed concurrently on the same day on all subjects. Sweat chloride was interpreted as normal (<40 mmol/L), borderline (40–59 mmol/L), or abnormal (≥60 mmol/L).10 NPD was performed as described by Knowles et al,19 by a single operator masked to other test results. The change in CFTR-mediated chloride diffusion following chloride-free and isoproterenol perfusion (ΔCl-free+Iso) was used as the primary diagnostic parameter. As previously described, the reference range for NPD was based on the range of overlap between reference non-CF and CF individuals: ΔCl-free+Iso normal (<−12 mV), intermediate (−12 to −7.7 mV), and abnormal (>−7.7 mV).18
CFTR genotyping
All subjects, including reference CF subjects with one or two unidentified CFTR mutations, underwent genotyping by multiplexed heteroduplex analysis followed by sequencing of identified fragments.20 Large deletions were detected using established conditions.21 ,22 Subjects who had no or one identified mutation underwent complete sequencing of all exons and adjoining introns.
Statistical analysis
Concordances between the sweat test and NPD were examined using observed agreement (calculated as the number of patients with the same diagnosis divided by the total number of patients) and weighted Cohen's kappa statistics. Kappa values range from –1 (complete disagreement) to 1 (perfect agreement), with 0 corresponding to agreement by chance alone. Because they condition on the marginal probabilities, they are considered conservative estimates of agreement. Kappa values were classified as: <0.20=poor; 0.21–0.40=fair; 0.41–0.60=moderate; 0.61–0.80=good; 0.81–<1.00=excellent.23
Results
Subject characteristics
Among 208 subjects with single-organ manifestations of CF, six were excluded (four RESP and two PANC) because NPD was not successful, resulting in 202 patients (68 RESP, 42 PANC and 92 AZOOSP). Eighty-six per cent were European Caucasians (see supplementary material, available online only). The majority of RESP and PANC subjects were women. The reference group included 104 healthy controls, 52 obligate heterozygotes, 64 CFPS and 43 CFPI subjects (table 1).
Subject characteristics and outcomes of sweat chloride and NPD testing
CFTR genotyping
There were 70 (34.7%), 41 (20.3%) and 91 (45.0%) undiagnosed subjects with none, one and two CFTR mutations, respectively. The number and frequency of identified mutations varied considerably between phenotypes (table 2). Based on the original 23 CF-causing mutations,10 only seven (3.5%) were diagnosable as CF, all of whom were diagnosed by sweat testing. Using CFTR2's expanded list of 122 mutations, 10 (4.9%) more subjects became diagnosable by genotyping, giving a total of 17 (8.4%) patients. Furthermore, diagnosis by genotype varied by phenotype: none in PANC, one of 68 (1.5%) in RESP, and nine of 92 (9.8%) among AZOOSP patients. In short, genotyping could not establish or exclude the diagnosis of CF in 74 of 91 (81.3%) with two CFTR mutations. Forty-seven of 91(51.7%) subjects with two CFTR mutations carried at least one mutation of varying clinical consequence: 44/91 (48.4%) carried a CF-causing mutation together with a mutation of varying clinical consequence, while three (3.3%) carried mutations of varying clinical consequence on both alleles. Of the 10 additional subjects with two CF-causing mutations designated by CFTR2, the diagnosis of CF could also be established by at least one abnormal ion channel measurement. Sweat testing alone missed three of 10 patients (one normal and two borderline results) while NPD testing alone missed two patients (both borderline results).
Breakdown of CFTR mutations in all subjects
All study subjects with no or one CFTR mutation (including healthy controls and heterozygotes) underwent extensive genotyping. No healthy controls or heterozygotes carried two CF-causing mutations, but a second mutation of unknown clinical consequence was identified in five obligate heterozygotes. CFTR2 increased the number of CFPI and CFPS patients fulfilling the diagnostic criteria for CF by genotype alone from 37 (86%) to 39 (90.7%) and 18 (28.1%) to 29 (45.3%), respectively.
Sweat chloride
Figure 1A demonstrates the variability of sweat chloride concentration within each group and the relationship (similarities and differences) among the groups. Among reference subjects, there was a spectrum and contiguity of increasing sweat chloride concentration from healthy controls and heterozygotes at one extreme to CFPI at the other. Among undiagnosed symptomatic individuals there was a similar wide range of sweat chloride measurements and sweat chloride values increased according to the number of mutations.
Thirty-five (17.3%), 46 (22.8%) and 121 (59.9%) undiagnosed patients had abnormal, borderline and normal sweat chloride results, respectively (table 1). Patients with AZOOSP had the highest proportion of subjects with borderline sweat chloride (36.9%), as compared to RESP (11.8%) and PANC (9.5%) subjects. Although none of the healthy controls and obligate heterozygotes had abnormal sweat chloride measurements, four (3.8%) and 11 (21.2%) had borderline results, respectively. All CFPI subjects had abnormal sweat chloride concentrations. However, only 41 (64%) CFPS subjects had abnormal sweat tests. Twelve (18.8%) and 11 (17.2%) CFPS subjects had normal and borderline sweat chloride concentrations.
Nasal potential difference
The wide spectrum of CFTR dysfunction noted on sweat testing was also observed with NPD (figure 1B). Similarly, all groups except CFPI had subjects with borderline NPD measurements. Among undiagnosed symptomatic patients, fewer patients had borderline NPD results than with sweat testing (18.8% vs 22.8%) (table 1). Thus, NPD could establish a diagnosis of CF in 67 (33.2%) subjects compared with 35 (17.3%) by sweat chloride.
Test concordance
According to the kappa statistic, there was a ‘moderate’ level of agreement between sweat chloride and NPD (table 3) among the RESP subjects, while among PANC and AZOOSP subjects, the levels of agreement were ‘poor’ and ‘fair’, respectively. Observed agreement for RESP, PANC and AZOOSP were 65%, 55% and 44%, respectively. Subanalysis was then performed after excluding all undiagnosed subjects with borderline results for either test. This analysis reflects discordance between the sweat test and NPD in subjects with clear-cut normal or abnormal diagnostic results (sweat test normal but NPD abnormal, and vice versa). Completely discrepant diagnostic outcomes (sweat chloride vs NPD) were present in 14%, 33% and 28% of RESP, PANC and AZOOSP subjects, respectively.
Concordance analysis between sweat chloride and NPD in all subjects and after excluding subjects with borderline results
In view of different lower cut-offs for borderline sweat chloride between the American and European guidelines (40 vs 30 mmol/L), concordance analysis was repeated using the 30 mmol/L cut-off. There was no difference in the concordance level between 40 versus 30 mmol/L cut-offs (see supplementary material, available online only).
Further comparisons of the diagnostic outcomes are shown in table 4 and figure 2. Among 121 undiagnosed subjects with normal sweat chloride, 24 (19.8%) had abnormal NPD results. Only 24 of 35 (68.6%) subjects with abnormal sweat chloride values had abnormal NPD results. Seven (20%) with abnormal sweat chloride values had normal NPD results.
Comparison of sweat chloride and NPD outcomes

{kind=link}
{kind=link}
Sweat chloride measurements plotted against NPD (ΔCl-free+Iso) for individual subjects with a single-organ manifestation of CF. The reference ranges for both tests are illustrated to identify patients with normal, borderline or abnormal values for each test, and demonstrate the discordance in diagnostic test outcomes (eg, bottom left: normal sweat chloride and NPD values; top right: abnormal sweat chloride and NPD; bottom right: normal sweat test but abnormal NPD; top left: abnormal sweat test but normal NPD. CF, cystic fibrosis; cross (+), subjects who presented with a single organ CF phenotype and one mutation; NPD, nasal potential difference; open circle (o), subjects who presented with a single organ CF phenotype and not identified with any CFTR mutation; solid circle (•), subjects who presented with a single organ CF phenotype and two mutations.
Discussion
The diagnosis of CF should be made with a high degree of certainty because it carries significant medical, financial and psychosocial implications. While the diagnosis of CF is usually established by the traditional sweat test alone in the majority of infants and young children, older children and adults presenting with single-organ manifestations suggestive of CF exhibit a wide overlapping spectrum of CFTR dysfunction. Therefore, the diagnosis of CF may be difficult to establish or exclude18 in those who fall within the ‘grey zone’ of the CFTR spectrum. This underlies the importance of having insight into the utility and limitations of diagnostic algorithms, various diagnostic tests, as well as knowledge of the spectrum of clinical manifestations at different ages.
In this study, the sweat test reliably discriminated individuals at the extreme ends of the CFTR spectrum, as evidenced by sweat chloride concentrations among healthy controls and CFPI. However, sweat chloride is limited by the wide overlapping values between non-CF and CF subjects. Approximately 20% each of obligate heterozygotes and subjects with single-organ manifestations of CF had borderline sweat chloride results. This is not surprising considering the upper limit of sweat chloride concentration among healthy non-CF individuals over 10 years old has been observed to overlap into the borderline range, with the upper limit for ages 10–14, 15–19 and over 20 years being 47, 51 and 56 mmol/L, respectively.26
Although several consensus reports offer varied opinions concerning their emphasis and role, genotyping and alternative ion channel measurements of the nasal and rectal epithelium have been proposed to clarify the diagnosis of CF in individuals with borderline sweat tests.10 ,11 ,16 ,27 While genotyping had previously been limited by the fact that only 23 of the more than 1900 CFTR mutations were designated as CF causing, this study shows that despite the expansion of the number of disease-causing mutations with CFTR2, genotyping still provided the lowest diagnostic yield when compared to the sweat test and NPD. Furthermore, all subjects with single-organ manifestations of CF diagnosable by genotyping had at least one ion channel measurement (sweat test and/or NPD) within the diagnostic range. In contrast to RESP and PANC patients, a large subset (68.5%) of AZOOSP subjects was identified with two CFTR mutations and was associated with the highest diagnostic yield from the list of 122 CF-causing mutations. A possible explanation is that among all the CFTR-affected organs, the vas deferens is most dependent on CFTR for ion transport and fluid secretion.28
The NPD measurements also demonstrated a wide spectrum of CFTR dysfunction with an overlapping ‘grey zone’ between non-CF and CF individuals. Among the three diagnostic tests, NPD identified the largest number of patients diagnosable with CF with fewer borderline outcomes. However, considerable discordance between the sweat test and NPD results is very concerning particularly when discordance remained after subjects with borderline results on sweat testing and/or NPD were excluded from the analysis. Discordance was lowest in RESP and highest in PANC subjects.
While the reasons for this discordance are unclear it may reflect variation in the penetrance of ion channel abnormalities in different CF-affected organs. Furthermore, ion channel measurements across different epithelia may be variably influenced by non-CFTR genetic and environmental factors. It seems plausible that discordant results will also be present between other ion channel measurements, for example, intestinal current measurement and β-adrenergic sweat secretion.27 ,29
There are several important questions. Is it appropriate to attribute a final diagnostic outcome based on sequential interpretation of diagnostic tests, which forms the basis of current European and American guidelines?10 ,11 As subjects can have a normal sweat test but abnormal NPD and vice versa, symptomatic individuals with a normal (or abnormal) sweat test may receive a spurious diagnosis because in such circumstances an NPD test is neither routinely recommended nor performed in clinical practice. In particular, do individuals with a normal sweat test plus abnormal NPD, or alternatively those with an abnormal sweat test but normal NPD have CF or not? In addition, as previously reported, a large number of the undiagnosed subjects in this study fulfil the criteria for a ‘CFTR-related disorder’.18
Despite the recent increased number of designated CF-causing mutations by CFTR2, genotyping continues to have limited diagnostic utility in challenging cases such as those presenting with a single-organ CF-like phenotype at a later age. There are several plausible explanations. The majority of newly identified disease-causing mutations by CFTR2 have severe phenotypic consequences, which are associated with clearly abnormal sweat chloride concentrations and confer the PI phenotype. Hence, most patients carrying these mutations could be identified by a simple sweat test alone. Second, half of the subjects with at least one or two identified CFTR alleles carried mutations designated by CFTR2 as having ‘varying clinical consequences’. As the majority of these mutations are associated with milder dysfunction, it is unsurprising that a large subset of patients with single-organ manifestations of CF were found to carry these mutations. Third, as most of the mutations reported in the CF mutation database (http://www.genet.sickkids.on.ca) are missense mutations, CFTR2 was neither able to assign a disease-causing nor a benign designation. Genotype interpretation may also be limited in non-European Caucasian groups, as common mutations in these groups have not been assessed by CFTR2. Nonetheless, genotyping may play an important role in situations in which sweat testing cannot be performed (eg, unavailability or prenatal diagnosis) or in situations in which disease causing mutations are associated with a normal or borderline sweat test or NPD. Nevertheless, the clinical utility of genotyping is expected to improve as more information regarding a greater number of specific mutations is obtained.
The role of in silico tools to predict the functional consequences of rare CFTR mutations has also been evaluated.30 ,31 While in silico tools may provide insight into the potential pathogenicity of rare mutations, they were shown to predict the clinical severity of missense mutations with known clinical consequences poorly, raising considerable doubt over their diagnostic role in mutations with variable or unknown clinical consequences.32
A diagnosis of CF in RESP patients has important clinical implications. However, follow-up should also be offered to PANC and AZOOSP patients with CFTR dysfunction due to the risk of disease development in other organs (eg, pulmonary disease). Subclinical pulmonary disease has been reported in AZOOSP men with two CFTR mutations and CFTR dysfunction, but due to a lack of longitudinal studies long-term pulmonary outcomes are unknown.33 Respiratory disease at/after CF diagnosis has also been reported in patients with pancreatitis.34 In addition, there may be a future role for CFTR-assist therapies in patients with pancreatitis.3 The female preponderance among RESP and PANC subjects is consistent with previous reports.2 ,35
This study has several strengths. We prospectively ascertained a large number of well-characterised older subjects from three single-organ phenotypes and who represented a wide spectrum of CFTR dysfunction. Furthermore, the findings on discordance between the sweat test and NPD were based on concurrent testing. The major limitations include the lack of longitudinal clinical monitoring and repeat ion channel testing. For practical and analytical reasons (ie, avoiding the need for separate visits and the complications of incomplete recall for a second test) we opted to perform each test concurrently on a single occasion. To obtain meaningful insight into the natural history of disease among older subjects with single-organ manifestations, prospective monitoring of a large sample over several decades is essential. Although approximately 5% of mutations may be missed by the genotyping methods used, we are confident that all 122 mutations listed by CFTR2 would have been identified.
In conclusion, the diagnosis of CF or its exclusion is not always straightforward and may remain elusive even with comprehensive evaluation, particularly among individuals who present at an older age with single-organ manifestations of CF. Considerable diagnostic uncertainty remains because many of these patients have ‘borderline’ and/or overlapping CFTR-mediated ion channel measurements, and there is considerable discordance between sweat testing and NPD. Finally, the diagnostic yield of CFTR2's expanded list of disease-causing mutations remains somewhat limited.
Acknowledgments
The authors would like to thank the many research subjects who gave up time to participate in the project and acknowledge the support and assistance of Louise Taylor, Susan Carpenter, Thora St Cyr, Rachel Paul, Debbie Ryan, Lenny Chong, Leia Spencer, Xiao-Wei Yuan, Qiuju Huang, Satti Beharry and Wan Ip.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors CYO and PD contributed to the study design, data acquisition, statistical analysis and interpretation and drafting of the manuscript. LE, SM, TG, KJ, PK, MS, JZ, RD, PNR, LS and ET contributed to data acquisition and critical revision of the manuscript. AD contributed to data analysis, statistical analysis and critical revision of the manuscript. All authors approved the final version of the manuscript. PD is the guarantor of the manuscript.
-
Funding PD and ET were supported by research grants from Cystic Fibrosis Canada (formerly Canadian Cystic Fibrosis Foundation) and Genome Canada through the Ontario Genomics Institute as per research agreement 2004-OGI-3-05, the Ontario Research Foundation and from the Lloyd Carr-Harris Foundation. CYO and TG were funded by the Cystic Fibrosis Canada fellowship awards and CYO received a Canadian Child Health Clinician Scientist Program career enhancement award. RD was funded by a Canadian Institutes for Health Research–Ontario Women's Health Council joint fellowship.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The research ethics board of The Hospital for Sick Children, St Michael's Hospital and Mount Sinai Hospital gave authorisation for the study.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Chest clinic
- Airwaves