Article Text

Abstract
Background Idiopathic pulmonary fibrosis is the most devastating fibrotic diffuse parenchymal lung disease which remains refractory to pharmacological therapies. Therefore, novel treatments are urgently required. Protease-activated receptor (PAR)-1 is a G-protein-coupled receptor that mediates critical signalling pathways in pathology and physiology. Bleomycin-induced lung fibrosis has been shown to be diminished in PAR-1-deficient mice. The purpose of this study is to investigate whether pharmacological PAR-1 inhibition is a potential therapeutic option to combat pulmonary fibrosis.
Methods Pulmonary fibrosis was induced by intranasal instillation of bleomycin into wild-type mice with or without a specific PAR-1 antagonist (ie, P1pal-12, a pepducin that blocks the PAR-1/G-protein interaction). Fibrosis was assessed by hydroxyproline analysis, immunohistochemistry, quantitative PCR and western blot for fibrotic markers expression.
Results We first show that P1pal-12 effectively inhibits PAR-1-induced profibrotic responses in fibroblasts. Next, we show that once daily treatment with 0.5, 2.5 or 10 mg/kg P1pal-12 reduced the severity and extent of fibrotic lesions in a dose-dependent manner. These findings correlated with significant decreases in fibronectin, collagen and α smooth muscle actin expression at the mRNA and protein level in treated mice. To further establish the potential clinical applicability of PAR-1 inhibition, we analysed fibrosis in mice treated with P1pal-12 1 or 7 days after bleomycin instillation. Interestingly, when administered 7 days after the induction of fibrosis, P1pal-12 was as effective in limiting the development of pulmonary fibrosis as when administration was started before bleomycin instillation.
Conclusions Overall, targeting PAR-1 may be a promising treatment for pulmonary fibrosis.
- Idiopathic Pulmonary Fibrosis
- Interstitial Fibrosis
Statistics from Altmetric.com
Key messages
What is the key question?
-
Is pharmacological protease-activated receptor (PAR)-1 inhibition a potential therapeutic option to combat pulmonary fibrosis?
What is the bottom line?
-
The cell penetrating PAR-1 antagonist P1pal-12 inhibits PAR-1-induced profibrotic effects in vitro and limits pulmonary fibrosis in preclinical animal experiments.
Why read on?
-
Targeting PAR-1 may be a promising treatment option for pulmonary fibrosis.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most frequent diffuse fibrosing lung disease of unknown aetiology.1 ,2 The prognosis of IPF is devastating with a 20–30% survival rate at 3–5 years after diagnosis and a mortality rate that exceeds many types of cancer. Since treatment approaches for IPF are limited, considerable effort has been made to reveal the underlying mechanisms allowing novel treatments to prolong survival.2 The molecular mechanisms underlying the pathogenesis of IPF are far from understood. However, current paradigms postulate that the development of fibrosis involves repeated epithelial cell injury leading to aberrant wound healing responses. Consistently, a hallmark of IPF is the presence of fibroblastic foci with differentiated fibroblasts which show myofibroblast phenotypes and secrete extracellular matrix (ECM) proteins that form depositions which subsequently establish fibrotic lesions.1–3
Protease activated receptors (PARs) are seven transmembrane domain receptors that belong to the family of G-protein-coupled receptors (GPCRs).4 In contrast to other GPCRs, PAR-1 activation involves proteolytic cleavage of the receptor by serine proteases. PAR-1 was originally identified on human platelets and its best known agonist is thrombin,5 although other ligands like FXa and certain matrix metalloproteinases have been described.6 ,7 Proteolytic activation of PAR-1 reveals a tethered ligand with a new N-terminus. Once irreversibly activated, PAR-1 initiates the recruitment of Gα subunits, Gq, Gi and G12/13. Together with Gβγ they trigger several downstream signalling events, which contribute to a striking range of pathophysiological functions.5 ,8 ,9 In addition to proteinases, PAR-1 can be activated by synthetic agonist peptides, which are designed according to the sequence of the cleaved N-terminus.
Accumulating evidence indicates that PAR-1 induces multiple processes that may promote pulmonary fibrosis. PAR-1 modulates mitogenesis and angiogenesis, alters lung vascular permeability, stimulates fibroblast migration, proliferation, ECM synthesis, and enhances inflammation in the pulmonary epithelium.10–13 In line, PAR-1-deficient mice are protected from bleomycin-induced acute lung inflammation and pulmonary fibrosis.14 The attenuated fibrotic response in PAR-1-deficient mice was associated with a reduction in the total collagen content in the lung and with decreased levels of proinflammatory and profibrotic mediators like interleukin (IL)-6 and monocyte chemoattractant protein 1 (MCP-1). Furthermore, PAR-1 expression is highly increased within fibroproliferative and inflammatory foci in fibrotic human lung. Overall, PAR-1 may thus have a critical contribution in promoting pulmonary fibrosis and PAR-1 may be a promising target to combat the development and progression of IPF.
In recent years, several PAR-1 antagonists have been designed, among which pepducins seem to be most promising. P1pal-12 is a cell-penetrating pepducin derived from the third intracellular loop of PAR-1. Once inserted into the plasma membrane it is delivered to the PAR-1 intracellular surface, thereby interfering with the receptor/G-protein interaction.15 P1pal-12 was initially described to inhibit PAR-1-driven calcium fluxes and platelet aggregation.16 ,17 In addition, P1pal-l2 has been shown to block PAR-1-mediated extracellular regulated kinase (ERK) activation in vitro.18 More importantly, PAR-1-specific pepducins also showed promising in vivo effects. Indeed, treatment with a PAR-1-specific pepducin significantly attenuated the growth of mice xenograft breast tumours,19 whereas the same pepducin provided remarkable inhibition of lung tumour growth in nude mice.20 P1pal-12 thus seems a promising PAR-1 inhibitor for preclinical experiments.
Here, we hypothesised that targeting PAR-1 with P1pal-12 may limit the development and progression of pulmonary fibrosis and tested this hypothesis using a well established murine model of bleomycin-induced pulmonary fibrosis.
Materials and methods
Cells and reagents
Mouse embryonic NIH3T3 fibroblasts (ATCC, Manassas, Virginia, USA; CRL-1658) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. Cells were grown at 37°C in an atmosphere of 5% CO2. Unless indicated otherwise, cells were washed twice with phosphate-buffered saline and serum starved for 4 h before stimulation. Thrombin was from Sigma (St Louis, Missouri, USA), whereas PAR-1 agonist peptide (PAR-1-AP; H-SFLLRN-NH2) and P1pal-12 (palmitate-RCLSSSAVANRS-NH2) were from GL Biochem (Shanghai, China).
Western blot
Western blots were performed essentially as described before.21 For details, see the online supplementary method section.
Wound scratch assay
Scratch assays were performed essentially as described before.21 For details, see the online supplementary method section.
Animal model of pulmonary fibrosis
Ten-week-old wild-type C57Bl/6 mice were purchased from Charles River (Someren, The Netherlands). All procedures were performed in accordance with the Institutional Standards for Humane Care and Use of Laboratory Animals. Experiments were approved by the Animal Care and Use Committee of the Academic Medical Center Amsterdam.
Bleomycin (Sigma, St-Louis) was administered intranasally (1 mg/kg) under anaesthesia. In the dose-finding experiment, animals were instilled with 0.5, 2.5 or 10 mg/kg P1pal-12 30 min before bleomycin administration and subsequently once daily until the end of the experiment. In the delayed treatment experiment, mice were treated once daily with 2.5 mg/kg P1pal-12 starting 1 or 7 days after bleomycin instillation. In both experiments, 6% dimethyl sulfoxide (DMSO) in saline was administered as solvent control. Mice were sacrificed 14 days after bleomycin instillation, after which one lung was taken for histology and one was homogenised.
Cytokine/chemokine assays
Transforming growth factor (TGF)-β1 was measured with the Mouse TGF-β 1 DuoSet kit (R&D systems, Abingdon, UK). IL-6 and MCP-1 were measured using the BD Cytometric Bead Array Mouse Inflammation Kit (BD, Franklin Lakes, New Jersey, USA) following the manufacturer's instructions. Detection limits were 2.5 pg/ml for IL-6 and 20 pg/ml for MCP-1.
Hydroxyproline assay
Right lungs were homogenised after which samples were processed for hydroxyproline content analysis using the hydroxyproline assay kit (Sigma, The Netherlands) as per the manufacturer's instructions. For details, see the online supplementary method section.
(Immuno)Histological analysis
Histological examination and Ashcroft score were performed as described before.22 α-SMA (smooth muscle actin) staining was graded in a blinded fashion on a scale from 0 to 3 as described before.23 Pictures of collagen staining were taken to cover the entirety of all sections. Colour intensity of stained areas was analysed semi-quantitatively with ImageJ and expressed as percentage of the surface area essentially as described before.24 ,25 For details, see the online supplementary method section.
Quantitative PCR
mRNA expression levels were quantified by real-time PCR as indicated in the online supplementary method section.
Statistics
Statistical analyses were conducted using GraphPad Prism (GraphPad software, San Diego, California, USA). Comparisons between conditions were analysed using two tailed unpaired t tests for normally distributed data, otherwise Mann–Whitney analysis was performed. p<0.05 was considered significant.
Results
P1pal-12 inhibits PAR-1-mediated signalling pathways in fibroblasts
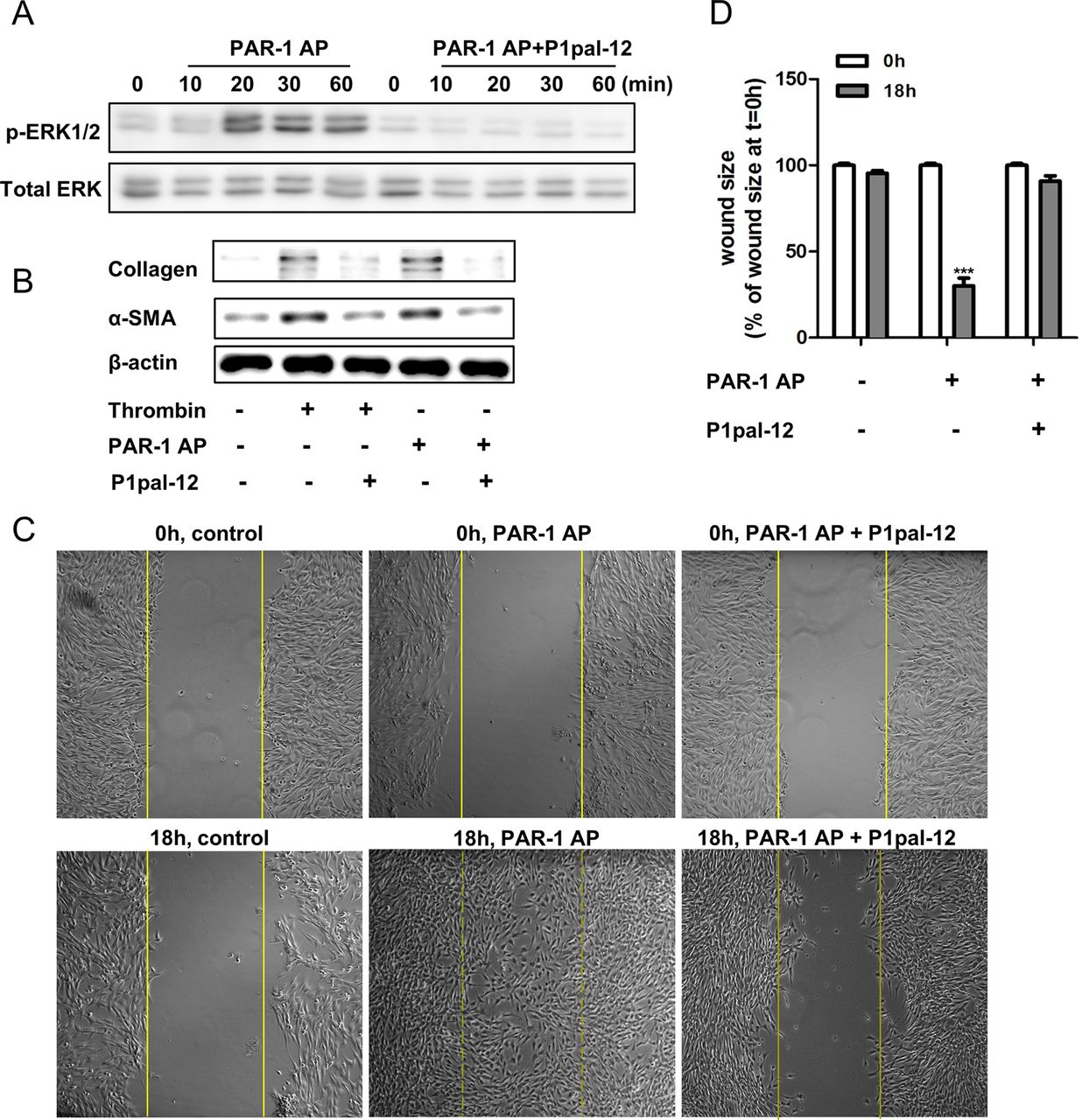
Several studies have shown that PAR-1 activation leads, among other things, to ERK phosphorylation, which is an important signalling pathway regulating fibroblast proliferation and migration.26 Pepducins have been shown to efficiently inhibit PAR-1-driven ERK phosphorylation in cancer cells.20 Importantly, however, pepducins may act in a cell-type-dependent manner.27 Therefore, we assessed whether P1pal-12 also inhibits PAR-1-driven ERK activation in fibroblasts. As shown in figure 1A, treatment of fibroblasts with 100 μM PAR-1-AP resulted in ERK1/2 phosphorylation. This PAR-1-AP-induced ERK phosphorylation was completely inhibited in 10 μM P1pal-12-pretreated cells. P1pal-12 thus effectively antagonises PAR-1-mediated signalling in murine fibroblasts.
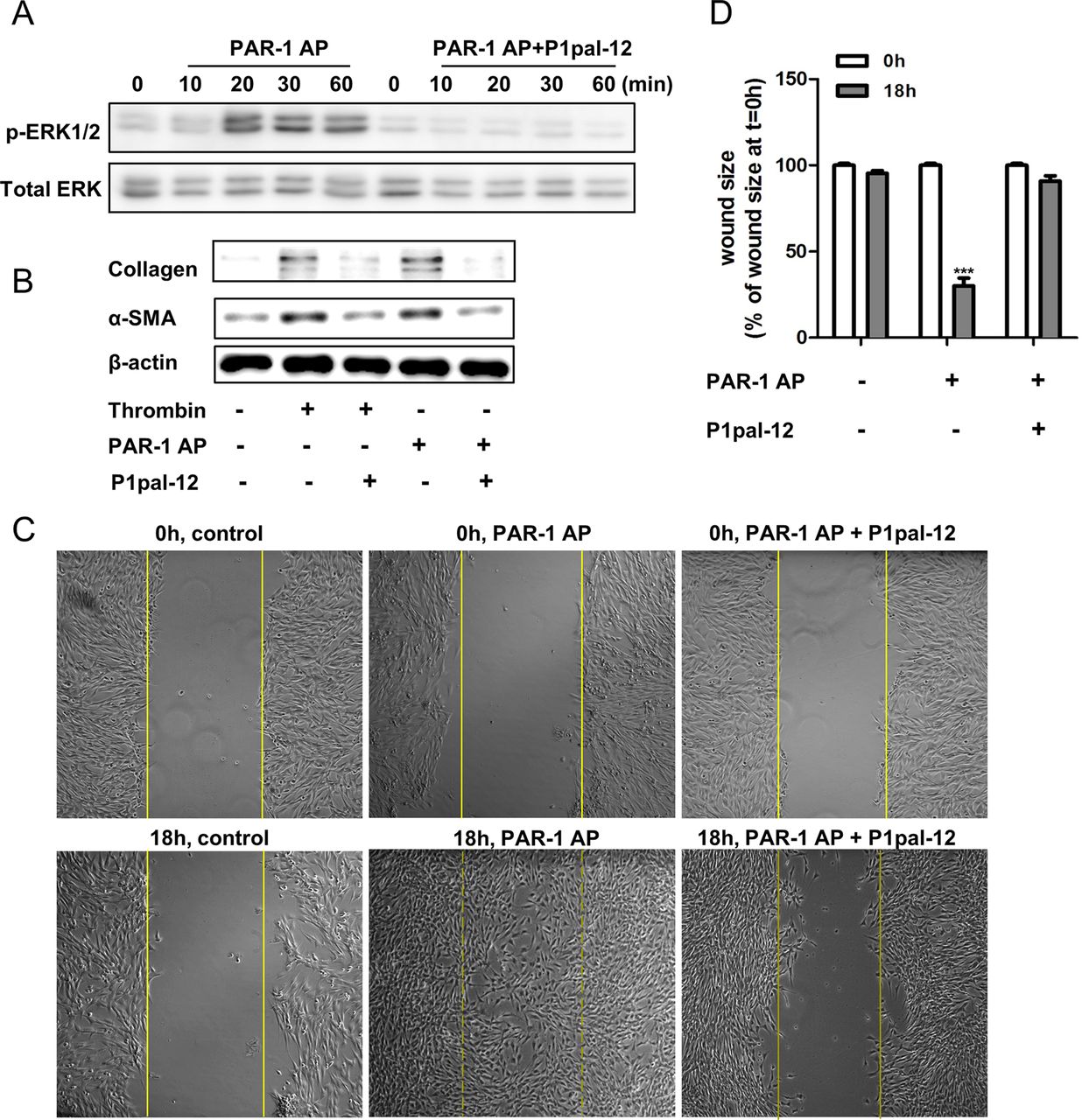
P1pal-12 inhibits protease-activated receptor (PAR)-1 induced profibrotic responses in fibroblasts. (A) Western blot analysis of extracellular regulated kinase (ERK) phosphorylation in NIH3T3 cells after stimulation with 100 μM PAR-1 agonist peptide (PAR-1-AP) in the absence or presence of P1pal-12 (10 μM). P1pal-12 was added 30 min before PAR-1-AP stimulation. Total ERK served as loading control. (B) Western blot analysis of α-SMA (smooth muscle actin) and collagen expression in NIH3T3 cells 24 h after stimulation with thrombin (1 U/ml) or 100 μM PAR-1-AP in the absence or presence of P1pal-12 (10 μM). P1pal-12 was added 30 min before thrombin or PAR-1-AP stimulation. β-actin served as a loading control. (C) Wound size of NIH3T3 fibroblast monolayers after treatment with either dimethyl sulfoxide (DMSO, control) or PAR-1-AP (100 μM) for 18 h in the presence or absence of P1pal-12. Cells were pre-incubated with 10 μM P1pal-12 for 30 min as indicated. Shown are photographs of representative microscopic fields. (D) Quantification of the results depicted in (C) as described in the Materials and methods section. Data are expressed as mean±SEM (n=6). ***p<0.001.
P1pal-12 inhibits PAR-1-dependent fibroblast differentiation, proliferation and migration
PAR-1 activation on fibroblasts has been demonstrated to induce several profibrotic processes like fibroblast migration, proliferation and differentiation.6 ,28 Consequently, we assessed whether P1pal-12 also antagonises these PAR-1-driven fibroproliferative responses. As shown in figure 1B, both PAR-1-AP (100 μM) and thrombin (1 U/ml) induced fibroblast differentiation and ECM synthesis as evident from increased α-SMA (hallmark of fibroblast differentiation) and collagen expression. This increased α-SMA and collagen expression was clearly downregulated by treatment with 10 μM P1pal-12. Importantly, TGF-β-induced collagen production is independent from P1pal-12 treatment (see online supplementary figure S1). Furthermore, we observed that PAR-1 stimulation induced fibroblast migration/proliferation in wound scratch assays, as evident from significantly smaller wound sizes (70–80%). Again, P1pal-12 treatment inhibited PAR-1-AP-induced wound closure (figure 1C,D). Overall, these in vitro experiments show that P1pal-12 effectively blocks the PAR-1-induced profibrotic effects of fibroblasts.
P1pal-12 dose dependently limits the development of pulmonary fibrosis in a murine bleomycin model
We next examined whether P1pal-12 limits fibrosis in a murine model of bleomycin-induced pulmonary fibrosis. To this end, mice were intranasally instilled with different concentrations of P1pal-12, after which the extent and severity of fibrosis was determined. As shown in figure 2A,B, bleomycin instillation induced extensive patchy fibrotic foci accompanied by a marked accumulation of inflammatory cells and increased deposition of ECM. Low-dose P1pal-12 treatment (0.5 mg/kg, figure 2C) marginally reduced bleomycin-induced fibrosis (about 10%) whereas intermediate (2.5 mg/kg) and high (10 mg/kg) P1pal-12 doses significantly reduced the severity of regional interstitial fibrosis (approximately 25% and 20%) and diminished the destruction of alveolar units (compare figures 2D,E with B). Quantification of the bleomycin-induced histological changes using the Ashcroft score shows that P1pal-12 treatment results in less severe fibrotic lesions (figure 2F).

P1pal-12 treatment affords protection against bleomycin-induced pulmonary fibrosis. (A–E, ×100) Representative H&E-stained lung tissue sections obtained 14 days after saline (A) or bleomycin instillation in mice treated with 0 mg/kg (B), 0.5 mg/kg (C), 2.5 mg/kg (D) or 10 mg/kg P1pal-12 (E). (F) Quantification of pulmonary fibrosis using the Ashcroft score in control mice and mice treated with P1pal-12. Data are expressed as mean±SEM (n=8 per group). (G–K, ×100) Representative pictures of α-SMA (smooth muscle actin) deposition in lung tissue sections obtained 14 days after saline (G) or bleomycin instillation of untreated (H) mice and mice treated with 0.5 mg/kg (I), 2.5 mg/kg (J) or 10 mg/kg (K) P1pal-12. (L) Quantification of α-SMA deposition as depicted in panels G–K. Data are expressed as mean±SEM (n=8 per group). *p<0.05. Dashed lines in the quantification figure represent solvent-treated controls.
It is well recognised that accumulation of α-SMA and ECM proteins (like collagen and fibronectin) can lead to organ-destructive remodelling,29 which also occur in fibrotic foci and are considered as hallmarks of IPF. To substantiate that P1pal-12 limits experimental fibrosis, we next analysed α-SMA expression immunohistochemically. Dramatic α-SMA expression was seen in focal fibrotic lesions after bleomycin instillation (compare figure 2G and H). Low-dose P1pal-12 treatment did marginally reduce α-SMA expression, whereas intermediate P1pal-12 doses significantly reduced (about 40%) α-SMA levels (figure 2I,J). Noteworthy, although the 10 mg/kg P1pal-12 dose also strongly reduced α-SMA levels; it did not reach statistical significance (figure 2K,L).
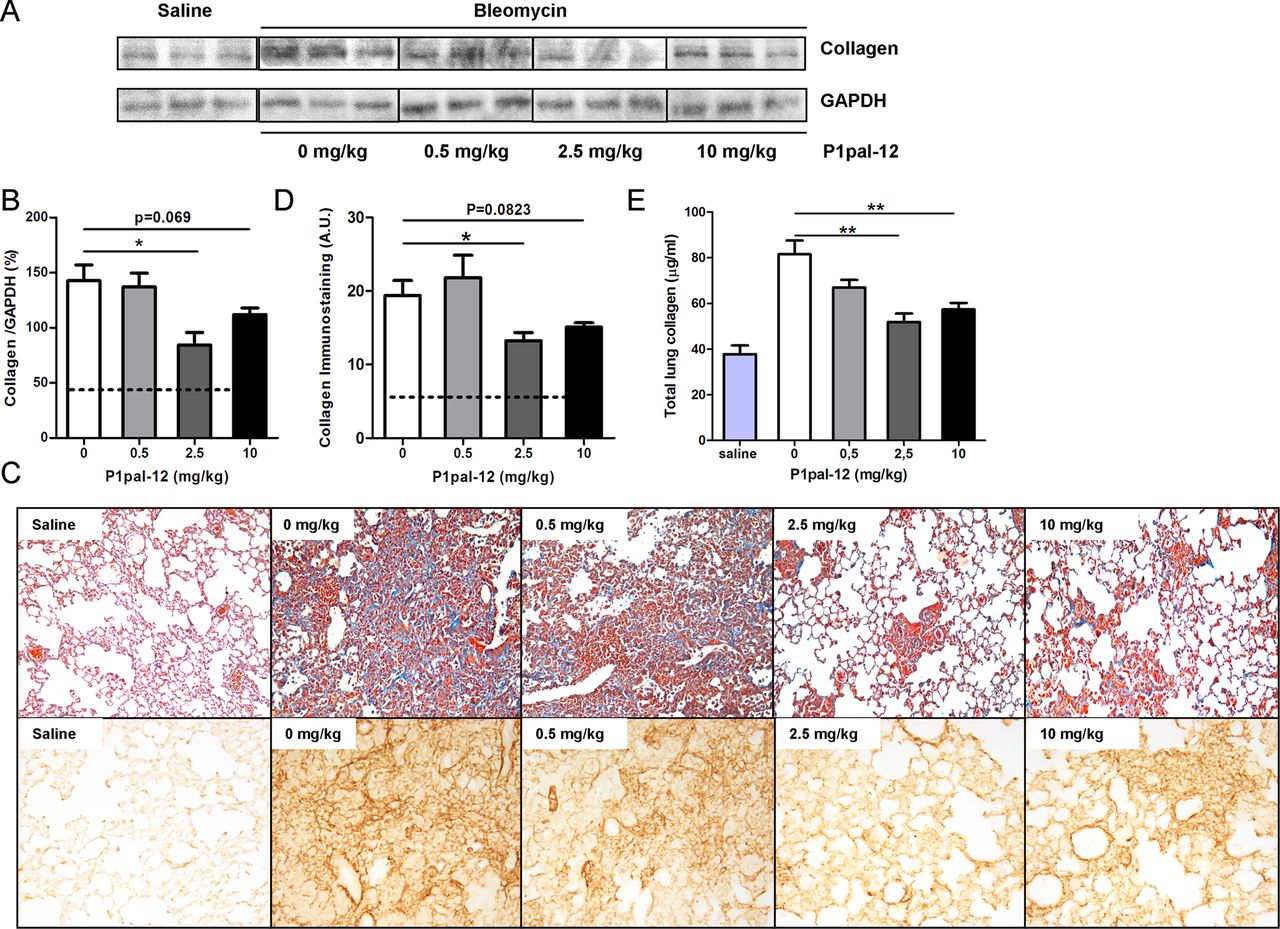
To confirm the inhibitory effect of P1pal-12 on bleomycin-induced fibrosis, we analysed collagen accumulation in the lung. Collagen levels in lung homogenates largely increased after bleomycin instillation. As shown in figure 3A,B, 2.5 and 10 mg/kg P1pal-12 administration considerably reduced collagen deposition in the lungs. In contrast, mice treated with 0.5 mg/kg P1pal-12 showed similar collagen deposition as mice not treated with P1pal-12. In line, Masson-trichrome and collagen stainings showed high collagen levels in bleomycin instilled mice, which were significantly reduced in mice treated with 2.5 or 10 mg/kg P1pal-12 (figure 3C,D). Finally, we confirmed these observations by analysing hydroxyproline content in right lung homogenates. As shown in figure 3E, collagen level (calculated according to the hydroxyproline content) increased almost threefold in bleomycin-treated mice compared with solvent controls. Importantly, collagen levels decreased 24.9±7.8%, 36.4±4.5% and 29.7±3.5% after treatment with 0.5, 2.5 and 10 mg/kg P1pal-12 respectively.

P1pal-12 treatment attenuates bleomycin-induced collagen deposition. (A) Western blot analysis of collagen expression in lung homogenates obtained 14 days after saline or bleomycin instillation in mice treated with different doses of P1pal-12. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control. Shown are three representative samples per condition from a group of eight. (B) Densitometric quantification of the results depicted in (A). Data are expressed as mean±SEM (n=8 per group; *p<0.05). (C) Representative pictures of Masson-trichrome (×100) and collagen-stained lung sections (×200) obtained 14 days after saline or bleomycin instillation in mice treated with 0–10 mg/kg P1pal-12. (D) Quantification of collagen immunostaining (semi-quantitative image analysis). (E) Collagen content in lung homogenates obtained 14 days after saline or bleomycin instillation in mice treated with different doses of P1pal-12. Dashed lines in the quantification figure represent solvent-treated controls.
Next, we analysed mRNA expression levels of α-SMA, collagen and fibronectin in the lungs. All these genes were highly expressed in response to bleomycin treatment and high as well as intermediate doses of P1pal-12 treatment strongly reduced their expression (figure 4A–C). Low-dose P1pal-12 treatment did not inhibit bleomycin-induced profibrotic gene expression.

P1pal-12 treatment reduces bleomycin-induced profibrotic gene expression and cytokine increases. Expression of α-SMA (smooth muscle actin) (A), collagen (B) and fibronectin (C) mRNA levels in lung homogenates obtained 14 days after saline or bleomycin instillation in mice treated with 0–10 mg/kg P1pal-12 was assessed by real-time reverse transcriptase PCR. Data are expressed relative to the amount of input RNA. Transforming growth factor (TGF)-β1 (D), interleukin (IL)-6 (E) and monocyte chemoattractant protein (MCP)-1 (F) levels in lung homogenates obtained 14 days after saline or bleomycin instillation in mice treated with 0–10 mg/kg P1pal-12. Data are expressed as mean±SEM (n=8 per group). *p<0.05, **p<0.01. Dashed lines represent solvent-treated controls.
Finally, we assessed TGF-β1 levels in lung homogenates as TGF-β1 is one of the most typical profibrotic mediators and is frequently overexpressed in fibrotic diseases.30 TGF-β1 levels increased in bleomycin-instilled untreated mice (twofold increase, figure 4D) compared with saline treated mice. The 2.5 mg/kg dose of P1pal-12 attenuated the TGF-β1 increase by about 50%. IL-6 and MCP-1 levels were also increased in bleomycin-instilled mice and again treatment with 2.5 mg/kg P1pal-12 significantly attenuated these increases by 65±3% and 36±3%, respectively. Treatment with 10 mg/kg P1pal-12 also led to a decrease in these cytokine levels, although it failed to reach statistical significance (figure 4E–F).
Overall, P1pal-12 limits bleomycin-induced pulmonary fibrosis, and the most significant effects were reached at the dose of 2.5 mg/kg. Therefore, we selected this intermediate dose for our subsequent studies.
Delayed treatment with P1pal-12 effectively limits pulmonary fibrosis progression in a murine bleomycin model of pulmonary fibrosis
After establishing that long-term treatment with p1pal-12 effectively limits pulmonary fibrosis, we next investigated whether delayed treatment starting after the initiation of fibrosis would still limit pulmonary fibrosis. Daily P1pal-12 (2.5 mg/kg) administration was started either 1 (inflammatory phase) or 7 (fibrotic phase) days after bleomycin instillation. As shown in figure 5A–C, lungs from mice not treated with P1pal-12 again showed severe fibrotic lesions induced by bleomycin treatment (more than fourfold increase compared with saline control). Interestingly, delayed P1pal-12 treatment reduced bleomycin-induced lung damage as evident from lower Ashcroft scores. However, scores of the 1-day delayed treatment group did not reach statistical significance (figure 5D). Consistently, α-SMA expression levels were increased in bleomycin-instilled lungs and these levels were significantly reduced in the 1-day and 7-day delayed treatment groups by 27±3.8% and 32 ± 8.0%, respectively (figure 5E–H).

Delayed P1pal-12 treatment effectively attenuates bleomycin-induced pulmonary fibrosis. (A–C) Representative H&E stained lung tissue sections obtained 14 days after bleomycin instillation in untreated mice (A) and mice treated with 2.5 mg/kg P1pal-12 from day 1 (B) or day 7 (C) after bleomycin instillation. (D) Quantification of pulmonary fibrosis using the Ashcroft score in untreated mice and mice treated with P1pal-12. Data are expressed as mean±SEM (n=8 per group). (E–G) Representative pictures of α-SMA (smooth muscle actin) deposition in lung tissue sections obtained 14 days after bleomycin instillation of untreated (E) mice and mice treated with 2.5 mg/kg P1pal-12 from day 1 (F) or day 7 (G) after bleomycin instillation. (H) Quantification of α-SMA deposition as depicted in panels E–G. Data are expressed as mean±SEM (n=8 per group). *p<0.05. Dashed line in the quantification figure represents solvent-treated controls.
Next, we determined pulmonary collagen levels and 1-day and 7-day delayed P1pal-12 treatment clearly reduced the expression of collagen (figure 6A,B). However, again, the effect of 1-day delayed treatment did not reach statistical significance. The decrease in collagen deposition by delayed P1pal-12 treatment was confirmed using collagen immunohistochemistry analysis and Masson-trichrome staining (figure 6C,D). In line, collagen content of bleomycin-instilled lungs increased by almost fourfold compared with saline control and this increase was attenuated by 34.5±3.8% and 40.7±3.7% in the 1-day and 7-day delayed treatment groups (figure 6E). Moreover, the decreases in α-SMA and collagen expression after delayed treatment were confirmed at the mRNA level (figure 6F–G), whereas fibronectin mRNA levels were also reduced after delayed treatment compared with P1pal-12 untreated controls (figure 6H).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Delayed P1pal-12 treatment attenuates bleomycin-induced collagen deposition and reduces gene expression of fibrotic markers. (A) Western blot analysis of collagen expression in lung homogenates obtained 14 days after bleomycin instillation in untreated mice and mice treated with 2.5 mg/kg P1pal-12 from day 1 or day 7 after bleomycin instillation. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control. Shown are three representative samples per condition from a group of eight. (B) Quantification of the results depicted in (A). (C) Representative pictures of Masson-trichrome (×100) and collagen-stained lung tissue sections (×200) obtained 14 days after bleomycin instillation in untreated mice and mice treated with 2.5 mg/kg P1pal-12 from day 1 or day 7 after bleomycin instillation. (D) Quantification of collagen immunostaining (semi-quantitative image analysis). (E) Collagen content in lung homogenates obtained 14 days after saline or bleomycin instillation in mice treated with different doses of P1pal-12. Expression of α-SMA (smooth muscle actin) (F), collagen (G) and fibronectin (H) mRNA levels in lung homogenates obtained 14 days after bleomycin instillation in untreated mice and mice treated with 2.5 mg/kg P1pal-12 from day 1 or day 7 post bleomycin instillation as assessed by real-time reverse transcriptase PCR. Data are expressed relative to the amount of input RNA. Data are expressed as mean±SEM (n=8 per group; *p<0.05, **p<0.01, ***p<0.001). Dashed lines in the quantification figures represent solvent-treated controls.
Overall, delayed treatment with P1pal-12 effectively limits the progression of bleomycin-induced pulmonary fibrosis. It is noteworthy, however, that P1pal-12 treatment starting 7 days after bleomycin instillation seems to be more effective compared with 1-day delayed treatment.
Discussion
IPF is the end result of a heterogeneous group of disorders with a devastating prognosis and very few therapeutic options.1 Interestingly, PAR-1 has been proven to play an important role in mediating proinflammatory and profibrotic effects in vitro, and preclinical experimental animal data showed that PAR-1 drives the progression of pulmonary fibrosis as mice that lack PAR-1 were protected against bleomycin-induced pulmonary fibrosis.14 In this study, we assessed whether treatment with a PAR-1 inhibitor limits experimental pulmonary fibrosis, and we show that pharmacological inhibition of PAR-1 with P1pal-12 indeed effectively limits pulmonary fibrosis in the murine bleomycin model. More specifically, we first show that P1pal-12 effectively blocks PAR-1 profibrotic effects in fibroblasts. Pre-incubation with P1pal-12 before PAR-1-AP or thrombin stimulation blocked ERK activation, diminished α-SMA expression and reduced proliferation/migration. Importantly, we were able to confirm these data in in vivo experiments by showing that bleomycin-induced fibrosis was significantly attenuated in mice treated with 2.5 and 10 mg/kg P1pal-12 administered once daily. Histological hallmarks of fibrosis and profibrotic protein and gene expression were significantly reduced after P1pal-12 treatment. Finally, we show that P1pal-12 still effectively limits pulmonary fibrosis when administration started 7 days after the induction of fibrosis by bleomycin instillation.
Interestingly, the observed effects of P1pal-12 are comparable to those observed in PAR-1-deficient animals, which confirms the efficacy of P1pal-12. In PAR-1-deficient mice14 and P1pal-12-treated mice, the severity of the fibrotic lesions was reduced by around one point in the Ashcroft score compared with untreated control mice. Moreover, collagen accumulation in the lung was reduced by around 55% in PAR-1-deficient mice, whereas P1pal-12 treatment reduced collagen deposition by approximately 40%. Finally, MCP-1 and TGF-β1 levels were increased in response to bleomycin instillation and these increases were diminished by 35% and 50% in PAR-1-deficient mice14 and by 36% and 50% in P1pal-12-treated animals for MCP-1 and TGF-β1, respectively. Overall, P1pal-12 treatment thus efficiently blocks PAR-1-driven pulmonary fibrosis.
Obviously, the fact that P1pal12 limits pulmonary fibrosis as efficiently as PAR-1 deficiency is scientifically interesting. However, more clinically relevant is the observation that when P1pal-12 treatment was started 7 days after the induction of fibrosis, it still significantly reduced fibrosis. The reductions in Ashcroft score, collagen deposition and profibrotic gene expression were similar to those observed in mice in which treatment was started before the induction of fibrosis. Inhibition of PAR-1 may thus be a promising therapeutic strategy for treating fibrosis, although future clinical studies are required to confirm this notion.
Two interesting findings of our study are that 2.5 mg/kg P1pal-12 seemed more effective in reducing fibrosis than the 10 mg/kg dose, and that initiation of treatment 7 days after bleomycin instillation was more effective than when treatment was started 1 day after the induction of fibrosis. Although we do not have a conclusive explanation for these surprising findings, it is tempting to speculate that the different dose-dependent and time-dependent effects are related to the dual role of PAR-1 in inflammation and fibrosis. In pulmonary fibrosis, PAR-1 is mainly considered to trigger inflammatory responses; however recent data suggest that PAR-1 also exerts anti-inflammatory functions.31 Therefore, the extent and/or timing of PAR-1 inhibition may influence the balance of its anti-inflammatory and proinflammatory properties. Alternatively, the slight differences observed may merely reflect the variability/inaccuracy associated with the analysis rather than any definable biological phenomenon.
When interpreting our data, several issues should be taken into consideration. First, although the bleomycin model is the best available model to study pulmonary fibrosis, it does not completely mimic the progression of fibrosis in patients with IPF. For example, the spontaneous resolution of fibrosis in this model fails to represent the irreversibility seen in patients with IPF. In addition, fibrosis develops fast in the bleomycin model, whereas it actually takes years to progress in patients.32 Second, we expressed our mRNA data as relative to the amount of input RNA as opposed to the more frequently used normalisation of gene expression against housekeeping genes. We opted for this approach because it is well known that expression of commonly used housekeeping genes is highly dependent on the experimental conditions33 ,34 and the use of total cellular RNA has been proposed as the best alternative for data normalisation.35 In agreement, we analysed several different ‘housekeeping genes’ (see online supplementary figure S2 for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), hypoxanthine–guanine phosphoribosyltransferase and 18S rRNA) and observed considerable variability between the different ‘house-keeping’ genes. Importantly, however, mRNA levels as expressed relative to the amount of input RNA do correspond with the histological scores and hydroxyproline analysis. Finally, we did observe a significant difference in IL-6 levels between untreated mice and mice receiving 2.5 mg/kg once daily. Such a difference in IL-6 was not shown in PAR-1-deficient animals. Most likely this is explained by the different time point at which IL-6 was measured (6 and 14 days after bleomycin instillation for PAR-1-deficient and P1pal-12-treated mice, respectively), although we cannot exclude small differences due to genetic PAR-1 deficiency versus pharmacological PAR-1 inhibition.
In conclusion, P1pal-12 significantly blocks PAR-1-induced profibrotic effects in vitro and inhibits bleomycin-induced pulmonary fibrosis in mice. Thus, targeting PAR-1 may be clinically relevant and may meet the urgent medical need for treating patients with pulmonary fibrosis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors CL, the guarantor for the overall content, conceived and designed the experiments, performed the experiments, analysed the data and wrote the paper; JWD performed part of the experiments; JD performed the animal experiments; MtB performed the animal experiments; JvdT performed part of the experiments and analysed the data; TvdP designed the experiments; KB conceived and designed the experiments, analysed the data and wrote the paper; CAS conceived and designed the experiments, analysed the data and wrote the paper. All authors critically revised the manuscript for important intellectual content and approved the currently submitted version. KB and CAS contributed equally.
-
Funding This work was supported by grants from TiPharma (T1-215-1) and the Netherlands Organisation for Scientific Research (016.136.167).
-
Competing interests None.
-
Provenance and peer review Not commissioned; internally peer reviewed.