Article Text

Abstract
Background Expression of the T-cell-associated chemokine receptor CCR8 and its ligand CCL1 have been demonstrated to be elevated in patients with asthma. CCR8 deficiency or inhibition in models of allergic airway disease in mice resulted in conflicting data.
Objective To investigate the effects of a selective small molecule CCR8 inhibitor (ML604086) in a primate model of asthma.
Methods ML604086 and vehicle were administered by intravenous infusion to 12 cynomolgus monkeys during airway challenge with Ascaris suum. Samples were collected throughout the study to measure pharmacokinetics (PK) and systemic CCR8 inhibition, as well as inflammation, T helper 2 (Th2) cytokines and mucus in bronchoalveolar lavage (BAL). Airway resistance and compliance were measured before and after allergen challenge, and in response to increasing concentrations of methacholine.
Results ML604086 inhibited CCL1 binding to CCR8 on circulating T-cells>98% throughout the duration of the study. However, CCR8 inhibition had no significant effect on allergen-induced BAL eosinophilia and the induction of the Th2 cytokines IL-4, IL-5, IL-13 and mucus levels in BAL. Changes in airway resistance and compliance induced by allergen provocation and increasing concentrations of methacholine were also not affected by ML604086.
Conclusions These results clearly demonstrate a dispensable role for CCR8 in ameliorating allergic airway disease in atopic primates, and suggest that strategies other than CCR8 antagonism should be considered for the treatment of asthma.
- Allergic lung disease
- Asthma
- Eosinophil Biology
- Lymphocyte Biology
- Pulmonary eosinophilia
Statistics from Altmetric.com
Key messages
-
What is the key question?
-
Does CCR8 inhibition efficiently ameliorate inflammation and airway function in a primate model of asthma?
-
What is the bottom line?
-
Inhibition of CCR8 with a small molecule antagonist did not result in improvements of lung inflammation and lung function, although complete inhibition of CCR8 on peripheral blood T-cells was achieved.
-
Why read on?
-
This study describes a dispensable role of chemokine receptor CCR8 in allergic airway inflammation and suggests that approaches other than CCR8 antagonism for targeting T-cells in asthma should be considered.
Introduction
Asthma is a chronic inflammatory disorder of the lungs characterised by airway obstruction, airway hyper-responsiveness and mucus hypersecretion, associated with episodes of wheezing and coughing.1 Increased numbers of perivascular and peribronchial eosinophils, mast cells and T helper 2 (Th2) effector cells along with the Th2 cytokines IL-4, IL-13 and IL-5 in the lung have been associated with asthma pathology.2–4 Allergen-specific activation of IgE-FceRI complexes on mast cells leads to the release of mediators including leukotrienes and histamine,5 ,6 as well as cytokines and chemokines, such as TNFα, IL-4, IL-13, CCL1 and CCL22.7–11
Chemokines constitute a family of small secreted proteins which bind to and activate specific seven transmembrane G-protein-coupled receptors (GPCR).12 The hallmark of chemokine receptor activation is induction of leukocyte migration along a chemokine gradient.12 Th2 effector cells preferentially express the chemokine receptors CCR3,13 CCR414 and CCR814 ,15 and migrate in response to their respective ligands CCL3, CCL17 and CCL1. Therefore, inhibition of chemokine receptors expressed on Th2 effector cells might be beneficial for the treatment of asthma.16
We have previously described an important role of mast cell-derived CCL1 and chemokine receptor CCR8 in the pathogenesis of allergic lung inflammation in mice,17 although others have reported a redundant role for CCR8 in similar models.18–20 A role for CCR8 in asthma pathogenesis is further supported by elevated levels of CCL1 and CCL1-expressing cells,17 ,21 ,22 as well as increased numbers of CCR8+ T-cells in asthma.16 ,17 ,22 In addition, CCR8 expression is mainly restricted to a subset of circulating human CD4 T-cells preferentially expressing the Th2 associated cytokines IL-4, IL-5, IL-9 and IL-13.15 ,22 Importantly, in humans, CCL1 is the only and specific ligand for CCR8, whereas in mice, CCR8 can also be activated by murine CCL8.23
In this study, we describe for the first time the consequences of CCR8 inhibition in a primate model of asthma using a small molecule CCR8 antagonist, ML604086.24 Although we were able to demonstrate >98% target inhibition by ML604086 on circulating T-cells, this inhibition did not ameliorate lung inflammation, Th2 cytokine production, mucus hypersecretion, methacholine (MCh)-induced airway hyper-responsiveness (AHR) and changes in lung function induced by inhaled allergen. Our data suggest that CCR8 antagonism was ineffective in ameliorating allergic airway disease and do not support CCR8 inhibition as a promising strategy for the treatment of atopic asthma.
Materials and methods
Animals and assignment
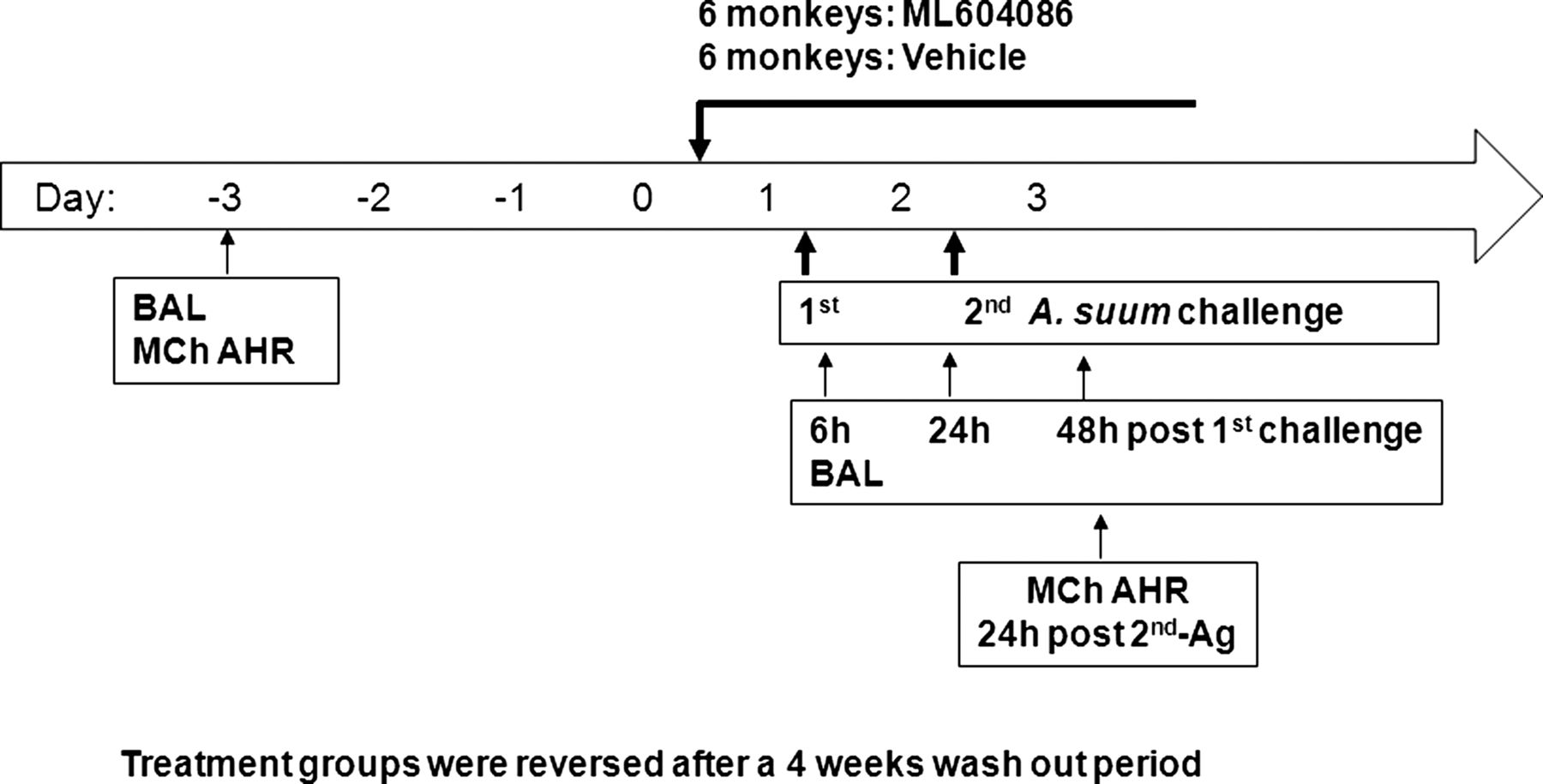
All animal procedures were conducted under protocols reviewed and approved by the Animal Care and Use Committee of the testing facility. Animals selected for this study, four male and eight female adult Macaca fascicularis, weighing between 3.0 kg and 5.0 kg and were obtained from the test facility's asthma colony (Charles River Laboratories, Worcester, Massachusetts, USA). All animals had previously demonstrated a positive bronchoconstrictor response to Ascaris suum antigen. These animals had been used in previous studies, and were deemed acceptable for use in this study. Animals were assigned to the study in a limited cross-over design consisting of two groups of six animals each. Group 1 animals received ML604086 during Session 1 and vehicle during Session 2. Group 2 received the vehicle during Session 1 and ML604086 during Session 2. Each challenge portion of the sessions was separated by at least 4 weeks. Data were summarised by combining animals from each treatment such that the same 12 animals are presented for both vehicle and ML604086 dosing sessions (figure 1).

Study design.
Dosing
The formulated ML604086 was prepared fresh on the first day of use for each session at a concentration of 1.31 mg/ml in 5% dextrose solution, sterile filtered, and stored at 22±5°C until dosing. Administration of ML604086 or vehicle (5% dextrose) was started on day 0 beginning 15 h prior to the first antigen challenge, and continuing until after collection of the final sample on day 3. Six animals were treated with ML604086 with a loading bolus dose of 1.038 mg/kg at a volume of 0.79 ml/kg over 5 min followed by a continuous infusion of 2.620 mg/kg/h at a volume of 2 ml/kg/h. Another six animals were treated with vehicle at the same rate for the same period. After 4 weeks washout period, the treatment groups were reversed. Dosing was accomplished through the use of a jacket and tether system, and the appropriate equipment to allow for continuous infusion.
Lung resistance and dynamic compliance (CDYN) measurements
Pulmonary function values were recorded throughout the challenge period using the Gould Ponemah computer system with pulmonary compliance and resistance modules (V.3.332), and are presented as the maximum per cent change from baseline (MaxΔ) values.
Methacholine (MCh) dose-response measurements
To determine bronchial hyper-responsiveness to MCh, an increasing concentration of MCh (0.1, 0.3, 1.0, 3.0, 10.0 and 30.0 mg/ml) was administered on day 3 for baseline measurement, and on day 3 (24 h post-2nd-antigen (Ag) challenge) until a strong positive response was seen (approximately 80% increase in lung resistance (RL) and/or approximately 50% decrease in CDYN), or until the highest concentration was administered.
Pharmacodynamic assay
To determine CCR8 inhibition by ML604086, blood was collected from vehicle or ML604086-treated monkeys at different time points. A 200 µl volume of blood was stained with 5 nM F-CCL1 (recombinant human CCL1 labelled with Alexa Fluor 647 obtained from Dictagene, Lausanne, Switzerland) at 37°C in a humidified incubator with 5% CO2/95% O2 for 30 min, followed by CD4 (L200) and CD45RA (5H9) (BD Biosciences, San Jose, California, USA) staining on ice for 30 min. Red blood cells were lysed with ammonium chloride lysis solution (StemCell Technologies, Vancouver, BC, Canada), and cells were washed once with Phosphate Buffered Saline (PBS) without Ca2+/Mg2+ (Invitrogen Corporation, Carlsbad, California, USA) containing 2% of Fetal Bovine Serum (FBS) (HyClone, Logan, Utah). The per centage of CCR8 in CD4 memory T-cells (CD4+ CD45RA-T-cell gate) was analysed by flow cytometry (BD FACS Calibur, BD). Predose blood from each monkey served as its own control. CCR8 inhibition by ML604086 was determined by the per centage of inhibition compared with control (100 nM human CCL1 (R&D Systems, Minneapolis, Minnesota, USA)), which was set to 100%.
Total and differential leukocytes analysis in bronchoalveolar lavage fluid
Total cell number per µl of bronchoalveolar lavage (BAL) fluid was determined using a Coulter Counter. To determine the per centage composition of each leukocyte type, the cells in BAL fluid from each time point were centrifuged onto slides using a Cytospin centrifuge, and stained with Diff-Quick for a differential cell count. Data were presented as absolute and relative cell numbers.
Statistical methods
A square root transformation was performed on cell counts, a rank transformation to analyte levels, and a logarithmic transformation to PC30. In order to test for changes in cells (counts and relative %s), analytes, PC30, and pulmonary function, prechallenge to postchallenge and treatment at each time point, as well as for differences between vehicle and ML604086, a linear model was applied accounting for a time (6, 24, or 48 h), random variation by animal within each treatment sequence (Group 1 or 2), and the effect of treatment by time. Goodness of fit was checked by adjusted R-squared and visually, including checks for normality of fitted residuals with normal quantile plots. The p value cutoff was set at 0.05, controlling the probability of three or more false positives to be ≤15%.
Results
CCR8 antagonist ML604086
In order to determine the inhibitory activity of select CCR8 antagonists for cynomolgus monkey (cyno; M fascicularis) CCR8, we cloned the CCR8 cDNA sequence from cyno peripheral blood mononuclear cells (PBMCs) (GenBank accession number: JX431563). Cyno CCR8 shared 94% and 72% amino acid identity with human and mouse CCR8, respectively (see online supplementary figure S1). Among the compounds tested, ML604086 (see online supplementary figure S2A) inhibited CCL1 mediated chemotaxis and increases in intracellular Ca2+ concentrations of cell lines stably expressing cyno CCR8 with IC50s of 1.3 µM and 1.0 µM, respectively (data not shown). Inhibition of cyno CCR8 by ML604086 could also be confirmed in cyno peripheral blood by measuring inhibition of fluorescently labelled CCL1 (F-CCL1) binding to CCR8+ CD4 memory T-cells (IC50=1.0 µM; see online supplementary figure S2B). ML604086 was selective against a panel of 40 G-protein coupled receptors (GPCR; Novascreen) when tested at 10 µM and 30 µM, and only moderately inhibited the serotonin receptor 5HT1a (30% at 10 µM and 70% at 30 µM). ML604086 competitively inhibited CCL1 binding to CCR8 and did not have any detectable agonistic activity, such as inducing CCR8 mediated chemotaxis. ML604086 was administered via intravenous infusion to 12 cyno monkeys to explore its effects on A suum mediated lung provocation in a cross-over study design (figure 1). Plasma levels of ML604086 ranged from 6.8 to 72.4 µM, 5 to 55-fold above the IC50 of ML605086, respectively. In three monkeys, we also evaluated ML604086 levels in BAL at 6, 24 and 48 h after the start of administration. ML604086 levels varied between 39 and 528 nM.
Inhibition of CCR8 on peripheral blood T-cells
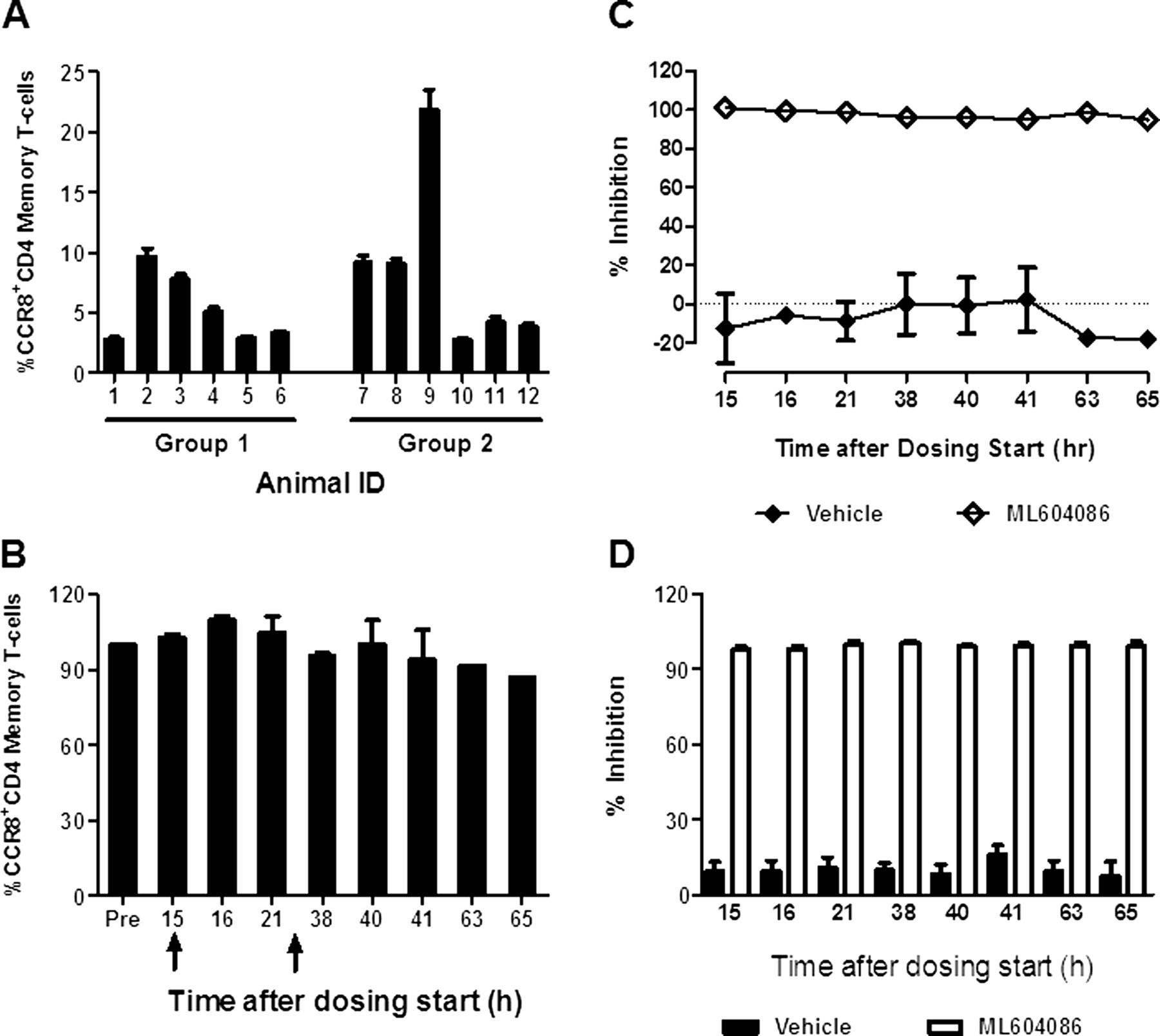
To monitor the engagement of ML604086 with CCR8 on circulating T-cells, we measured its ability to displace F-CCL1 binding. We first determined the per cent of CCR8 expressing CD4 memory T-cells prior to ML604086 or vehicle administration. Numbers varied between individual animals, ranging from 3% to 23% (figure 2A). Interestingly, the number of CCR8 expressing CD4 memory T-cells in each animal was quite stable and was not affected by allergen provocation (figure 2B). ML604086 completely inhibited the binding of F-CCL1 to CD4 T-cells (>98%) at any time point investigated, whereas, vehicle administration did not interfere with F-CCL1 binding (figure 2C). Average CCR8 inhibition in all animals receiving ML604086 or vehicle is summarised in figure 2D. Our data clearly demonstrate that ML604086 administration completely inhibited F-CCL1 binding to CCR8 expressed on CD4 T-cells at all times investigated, and that allergen challenge did not change the proportion of circulating CCR8+CD4 T-cells.

CCR8 inhibition in blood by ML604086. (A) % of CCR8+CD4CD45RA− T-cells/animal at baseline. Error bars represent SEM from two independent experiments. (B) Change in the % of CCR8+CD4CD45RA− T-cells for one representative animal during allergen challenge (arrows) compared with control (predose). Error bars represent SEM from two independent experiments. (C) Inhibition of F-CCL1 binding to CCR8+CD4CD45RA− T-cells by ML604086 or vehicle in comparison with control (100 nM CCL1 at predose) for one representative animal. Data represent mean±SEM from two independent experiments. (D) Mean of CCR8 inhibition by ML604086 in all 12 animals. Data represent the mean±SEM.
Lung inflammation, cytokines and mucus
Allergen challenge resulted in a substantial inflammatory response in BAL, starting at 6 h and increasing at 24 and 48 h postchallenge (figure 3A). Eosinophils were the dominant infiltrating cell type (approximately 60–70%; figure 3B,C), followed by increasing numbers of macrophages (figure 3D,E). Absolute numbers of neutrophils and lymphocytes remained largely unaffected (data not shown). Administration of ML604086 did not reduce the total number of infiltrating cells or reduce the number of BAL eosinophils and macrophages in comparison with vehicle (figure 3A–E). Next, we quantified cytokine levels in BAL by ELISA. Levels of IL-4 were elevated to statistically relevant levels 6 h after allergen provocation (figure 4A). IL-5 and IL-13 were below the limit of detection at baseline but increased significantly 6 h after allergen provocation (figure 4B,C). Levels of IFNγ did not exceed the limit of detection at all times investigated (data not shown). There was no significant change in cytokine levels between vehicle and ML604086 groups (figure 4A–C). A hallmark of allergic mucosal inflammation is goblet cell hyperplasia and associated mucus hypersecretion. We therefore measured mucus production in BAL using ELISA. Mucus levels were found to increase as early as 6 h (approximately 50 µg/mg total protein) and increased to approximately 100 µg/mg total protein at 48 h after antigen challenge (figure 4D). No differences between ML604086 and vehicle groups were found.
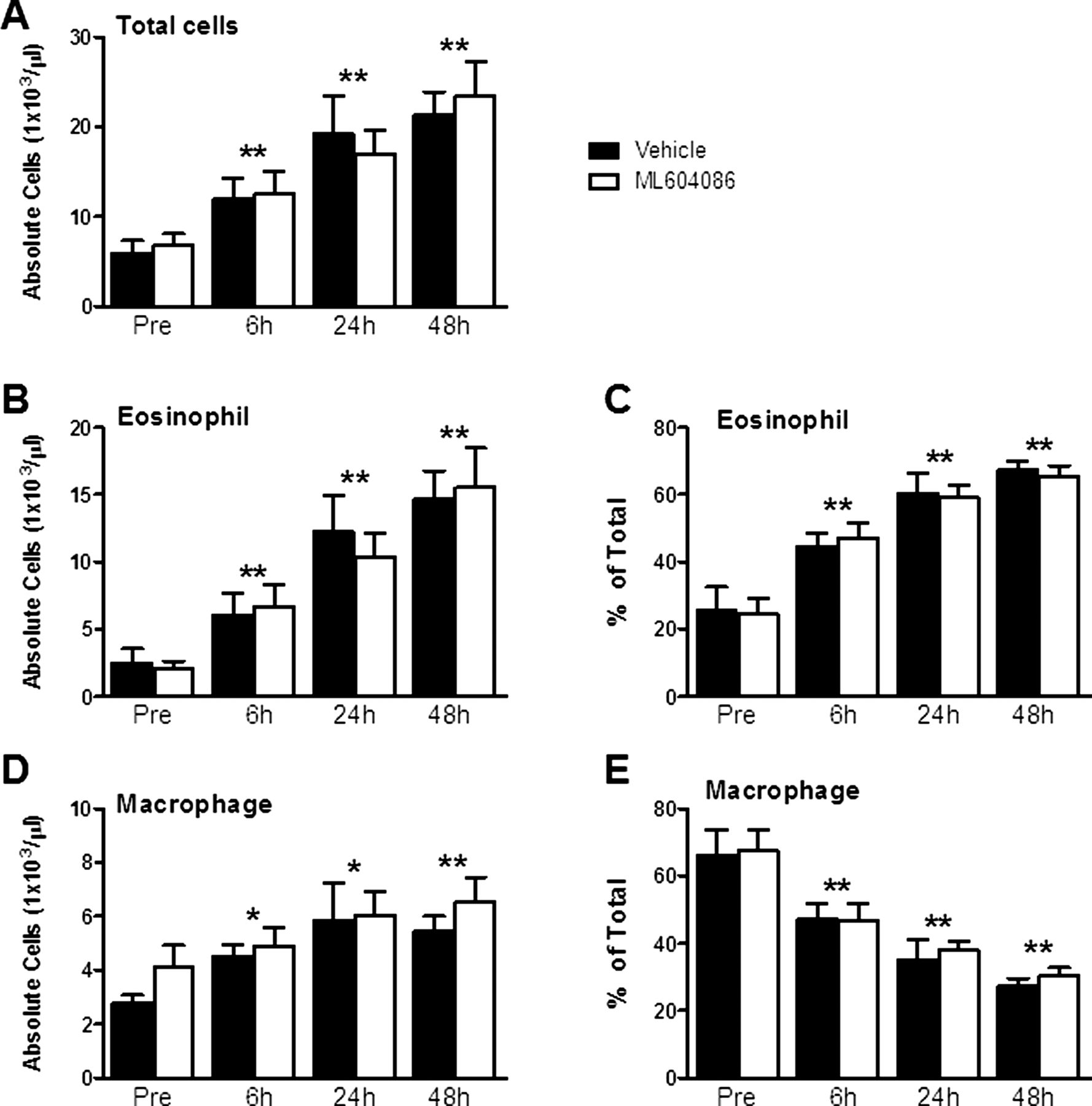
Cell counts in bronchoalveolar lavage. Absolute (A, B and D) and relative (C and E) cell counts of total cells (A), eosinophils (B and C) and macrophages (D and E). Data are presented as mean±SEM from all 12 animals. p Values different from pre, p<0.05 (*) and p<0.001 (**), were calculated by linear model analysis of cell counts or per cents accounting for the cross-over design (see Statistical methods). No statistically significant differences were found between both groups at any time point.

Bronchoalveolar lavage cytokines and mucin. Levels of IL-4 (A), IL-5 (B), IL-13 (C) and mucin (D) were quantified by ELISA and normalised to total protein levels. Data represent mean±SEM of all 12 animals treated with vehicle (solid bars) or ML604086 (open bars). p Values different from pre, p<0.05 (*) and p<0.001 (**), were calculated by linear model analysis of analytes accounting for the cross-over design (see Statistical methods). No statistically significant differences were found between both groups at any time point.
Pulmonary function
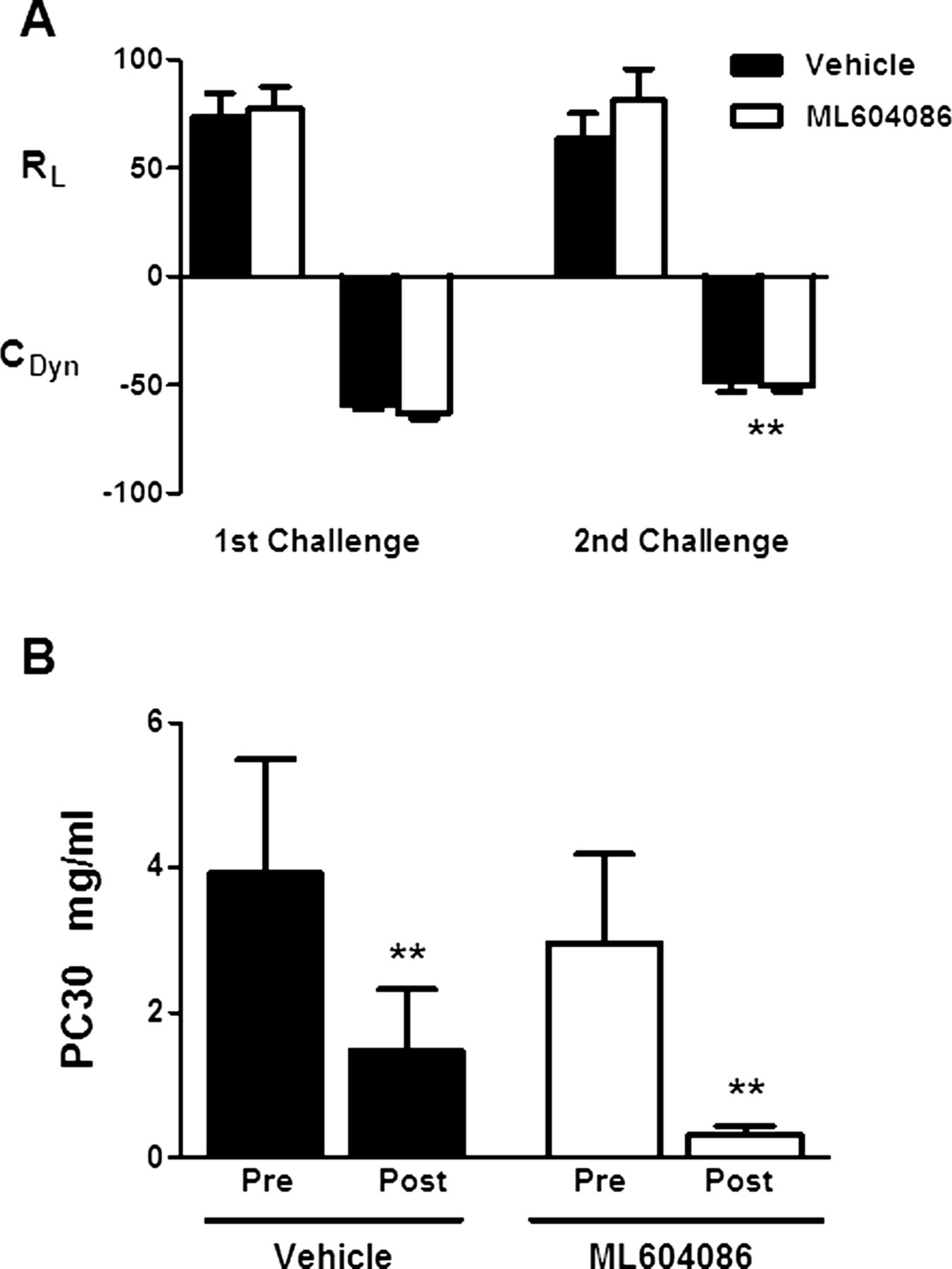
A suum antigen challenge resulted in a strong bronchoconstriction response in all animals. The mean increases from baseline in lung resistance (MaxΔRL) of vehicle-treated animals after the first and second allergen provocation were 74% and 64%, respectively. These values tracked with changes of −59% and −48% from baseline in dynamic compliance (MaxΔCDYN), respectively (figure 5A). Treatment with ML604086 did not result in any significant difference in lung resistance or compliance in comparison with vehicle-treated animals (figure 5A). To assess the effects of ML604086 treatment on AHR, animals were exposed to increasing doses of inhaled MCh. Animals in vehicle and ML604086-treated groups, showed a similar increase in RL with increasing doses of MCh. From these measurements, the MCh PC30 (the concentration of MCh resulting in a 30% change from baseline) values were calculated using non-linear regression analysis (figure 5B). Two animals were excluded from the PC30 analysis because an increase of 30% from baseline could not be achieved at the highest dose administered. PC30 values dropped in the ML604086 group from 2.968 mg/ml and 0.325 mg/ml on days −3 and 3, respectively (figure 5B), indicating a positive AHR. A similar increase in MCh sensitivity was found in the vehicle group with PC30 values dropping from 3.916 mg/ml on day −3 to 1.468 mg/ml on day 3 (figure 5B). In conclusion, treatment with ML604086 did not inhibit development of AHR to MCh following A suum antigen challenge.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pulmonary function. (A) Changes in pulmonary airflow resistance (RL) and dynamic compliance (CDYN) upon allergen challenge in vehicle (solid bars) or ML604806 (open bars) treated animals. Results are presented as mean±SEM of per cent change relative to baseline. (B) Airway hyper-responsiveness was determined measuring pulmonary airflow resistance (RL) with increasing doses of MCh. The concentration of MCh (mg/ml) resulting in a 30% change from baseline was calculated (PC30). Values are mean±SEM from 10 animals treated with vehicle (solid bars) or ML604086 (open bars). p Values different from first challenge (A) or pre (B), p<0.05 (*) and p<0.001 (**), were calculated by linear model analysis for the cross-over design (see Statistical methods). No statistically significant differences were found between both groups at any time point.
Discussion
Several lines of evidence suggest a role of the CCR8:CCL1 axis for the recruitment of Th2 cells to the asthmatic lung.16 ,17 ,22 However, conflicting data for a role of this axis using mouse models of allergic airway inflammation and difficulties to reliably determine CCL1 expression and CCR8+ T-cell numbers warrant additional studies aiming to delineate the relative importance of this pathway to asthma pathogenesis.25
Cynomolgus monkeys naturally allergic for the parasitic nematode A suum share significant lung pathology with atopic asthmatics, and are therefore considered valuable predictors for drug effects in humans.26 This notion has recently been supported by the beneficial effects of monoclonal antibodies inhibiting IL-1327 ,28 and IL-529–31 in asthmatics. In hindsight, exploration of the above pathways using allergen provocation in primate models of asthma has proven to be a powerful approach in predicting positive outcomes in clinical trials32–34 This is corroborated by early studies establishing the effects of well-known antiallergic and antihistaminic medications in primates.35 Investigations of the relative importance of the CCR8 pathway in A suum-elicited allergic airway inflammation in monkeys should, therefore, be of substantial predictive value for effects in patients with asthma.
Here we report for the first time the consequences of chemokine receptor CCR8 inhibition with a small molecule antagonist in A suum atopic monkeys. All monkeys used in this study reacted to A suum antigen challenge with an immediate bronchoconstriction response, and displayed increased AHR to inhaled MCh. In BAL, A suum challenge elicited a transient increase in infiltrating leukocyte numbers, in particular, eosinophils. As expected, and reported by others, allergen provocation did not result in elevated numbers of T-cells in BAL,32–36 but has been demonstrated previously to result in elevated numbers of activated tissue-resident CD4 T-cells and CCL1 expression in the lung in a similar model.37 T-cell accumulation and activation is further supported by elevated levels of the Th2 cytokines IL-4, IL-5 and IL-13 in BAL upon allergen provocation (our data). Interestingly, we found that challenge of the airways with allergen did not significantly change the numbers of circulating CCR8 expressing CD4 memory T-cells. These data may suggest that either CCR8 exerts its function mainly on tissue-resident T-cells by mediating T-cell activation and proliferation, or other chemokine receptors expressed on Th2 cells, such as CCR4, are involved in the recruitment of T-cells into the inflamed lung. Indeed, recent data demonstrate that in a humanised model of asthma CCR4 is critical for the recruitment and activation of human Th2 cells and subsequent airway eosinophilia, goblet cell hyperplasia and AHR.38 Our data in atopic monkeys further support that receptors other than CCR8 are important for orchestrating Th2 lung inflammation. CCR8 antagonism had no effects on allergen-induced induction of Th2 cytokines (IL-4, IL-5 and IL-13) in BAL and airway eosinophilia in comparison with vehicle administration. Consequently, CCR8 blockade did not affect mucus secretion, allergen-induced bronchoconstriction and AHR many of which have been shown to be mediated or enhanced by IL-13.39 To our disappointment, CCR8 blockade did not result in any beneficial effects. We believe that our study design provides strong evidence for such a conclusion. First, the cross-over design of the study allowed for the comparison of ML604086 with vehicle in all 12 monkeys used, eliminating possible intergroup differences resulting from the outbred nature and heterogeneity of animals. Second, through intravenous infusion administration of ML604086 we were able to achieve high plasma concentrations of ML604086 resulting in >98% inhibition of CCR8-lingand binding sites on circulating T-cells throughout the phase of allergen provocation. Although we cannot exclude incomplete inhibition of CCR8 expressed on tissue-resident T-cells, we believe that, based on the high plasma concentrations of ML604086, this explanation is quite unlikely. This conclusion is further supported by the detection of ML604086 in BAL of three selected animals (39–528 nM). These concentrations most likely underestimate actual concentrations in the lung due to the highly dilute nature of BAL.
Altogether, we conclude that lack of effects with ML604086 administration cannot be ascribed to incomplete target inhibition, but most likely is due to a dispensable role of chemokine receptor CCR8 in orchestrating allergic airway inflammation. We cannot rule out that CCR8-mediated T-cell recruitment and activation may play a role under circumstances in which innate immune system activation induced by viruses, bacteria or fungi, all of which have been shown to be associated with asthma pathology, contribute to airway inflammation. Because haematopoietic cells, including T-cells, express a variety of chemokine receptors on a single-cell level, and the promiscuity of chemokines for their receptors, a high level of redundancy has been proposed previously constituting an inherent problem for targeting chemokine receptors. Our data suggest that targeting other chemokine receptors than CCR8, such as CCR4, may hold larger promise for the treatment of atopic asthma.
Acknowledgments
The authors thank Dr Alison Humbles for critically reading the manuscript and insightful comments and Dr David Gold for his help with statistical analysis of the data.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors LW, TJJ, MD, WY, JCP, ELM, MRH, TO and RK have made substantial contributions to the conception and design, or analysis and interpretation of data. LW, MRH, TO and RK have drafted the manuscript or revised it critically for important intellectual content. LW, TJJ, MD, WY, JCP, ELM, MRH, TO and RK have approved the version of the manuscript for publication.
-
Competing interests None.
-
Provenance and peer review Not commissioned; internally peer reviewed.
-
Data sharing statement Additional unpublished data from the study are standard safety measurements (haematology, observatory assessments, etc) performed on all animals included in this study. The data are summarised in the form of a study report which is available to ELM and RK.