Article Text

Abstract
Background Viral infections are the most frequent cause of asthma exacerbations and are linked to increased airway reactivity (AR) and inflammation. Mice infected with respiratory syncytial virus (RSV) during ovalbumin (OVA)-induced allergic airway inflammation (OVA/RSV) had increased AR compared with OVA or RSV mice alone. Furthermore, interleukin 17A (IL-17A) was only increased in OVA/RSV mice.
Objective To determine whether IL-17A increases AR and inflammation in the OVA/RSV model.
Methods Wild-type (WT) BALB/c and IL-17A knockout (KO) mice underwent mock, RSV, OVA or OVA/RSV protocols. Lungs, bronchoalveolar lavage (BAL) fluid and/or mediastinal lymph nodes (MLNs) were harvested after infection. Cytokine expression was determined by ELISA in the lungs or BAL fluid. MLNs were restimulated with either OVA (323–229) peptide or RSV M2 (127–135) peptide and IL-17A protein expression was analysed. AR was determined by methacholine challenge.
Results RSV increased IL-17A protein expression by OVA-specific T cells 6 days after infection. OVA/RSV mice had decreased interferon-β protein expression compared with RSV mice. OVA/RSV mice had increased IL-23p19 mRNA expression in lung homogenates compared with mock, OVA or RSV mice. Unexpectedly, IL-17A KO OVA/RSV mice had increased AR compared with WT OVA/RSV mice. Furthermore, IL-17A KO OVA/RSV mice had increased eosinophils, lymphocytes and IL-13 protein expression in BAL fluid compared with WT OVA/RSV mice.
Conclusions IL-17A negatively regulated AR and airway inflammation in OVA/RSV mice. This finding is important because IL-17A has been identified as a potential therapeutic target in asthma, and inhibiting IL-17A in the setting of virally-induced asthma exacerbations may have adverse consequences.
- Allergic lung disease
- Asthma
- Respiratory Infection
- Cytokine Biology
Statistics from Altmetric.com
Key messages
What is the key question?
-
What is the role of IL-17A in airway inflammation and airway reactivity in a clinically relevant model of asthma exacerbation in which viral infection occurs during ongoing allergic airway inflammation?
What is the bottom line?
-
IL-17A negatively regulates airway reactivity and airway inflammation with respiratory syncytial virus infection during ongoing allergic airway inflammation.
Why read on?
-
IL-17A has been identified as a potential therapeutic target in asthma, and it is vital to understand the role of IL-17A in increased allergic airway inflammation and airway reactivity with viral infections during ongoing allergic airway inflammation.
Introduction
Asthma is characterised by increased lung inflammation, airway reactivity (AR) and mucus production. Viral infections trigger 80–85% of asthma exacerbations in children1 and 44% in adults.2 Most of these viral-induced asthma events occur in patients with allergic airway inflammation.3 Respiratory syncytial virus (RSV) is associated with many wheezing episodes and asthma exacerbations.4 However, the exact mechanisms by which viral infections cause asthma exacerbations remain unknown.
To determine the immunological mechanisms associated with viral-induced asthma exacerbations, we and others have previously developed mouse models of RSV strain A2 infection during ongoing ovalbumin (OVA)-induced allergic airway inflammation (OVA/RSV).5 ,6 In our model, 8 days after infection, OVA/RSV mice had the same AR as OVA mice and both groups were significantly increased compared with non-allergic uninfected mice (mock) or RSV-infected mice. However, 15 days after RSV challenge, OVA/RSV mice had significantly increased AR compared with OVA mice, in which AR had declined over time. This model closely mimics the heightened and prolonged AR that occurs with virally-induced asthma exacerbations in people. Interestingly, RSV strain A2 infection without allergic airway inflammation did not cause increased AR over mock mice at any time point.5
Interleukin (IL)-13, a Th2 cytokine, is a central mediator of AR and airway mucus production in allergic airway models. Lung IL-13 protein expression was significantly increased in OVA mice compared with mock mice,7 ,8 but lung IL-13 protein expression was significantly reduced in OVA/RSV mice compared with OVA mice.7 ,8 These findings suggest that IL-13 was not responsible for increased AR in OVA/RSV mice. However, OVA/RSV mice had a significant increase in lung IL-17A protein expression,8 a cytokine associated with severe asthma.9 ,10 Based on these findings, we hypothesised that IL-17A mediates increased AR and inflammation that occurs in OVA/RSV mice compared with WT mice.
IL-17A is produced primarily by Th17 cells and signals through a heterodimeric receptor composed of IL-17RA and IL-17RC subunits.11 It is associated with autoimmune diseases such as multiple sclerosis, rheumatoid arthritis and inflammatory bowel diseases, as well as protection against extracellular pathogens.11 ,12 IL-17A protein expression is also increased in bronchoalveolar lavage (BAL) fluid and bronchial biopsy tissue of patients with moderate to severe asthma.9 ,10
The role of IL-17A in allergic airway inflammation is conflicting. Mice deficient in IL-17RA failed to develop OVA-induced allergic airway inflammation.13 One potential reason was the inability of IL-17RA knockout (KO) mice to respond to IL-17E (also known as IL-25) which was required for allergic airway inflammation and increased IL-13 protein expression, eosinophil recruitment and AR.14 In the same study, Schnyder and colleagues also reported that exogenous instillation of rmIL-17A during OVA challenge decreased AR and eosinophil infiltration.13 IL-17A produced from γδ T cells also protected against AR.15 However, IL-17A was also reported to be important in the development of allergic airway inflammation. Adoptive transfer of OVA-specific Th17 cells or OVA-specific Th17 plus Th2 cells increased AR in BALB/c mice.16 ,17 Instillation of rmIL-17A into the airways also increased AR in mice with OVA-induced allergic airway inflammation.18 Taken together, these studies show that IL-17A both enhanced and inhibited AR in mouse models of allergic airway inflammation. The role of IL-17A in viral-induced AR and airway inflammation in the setting of underlying allergic inflammation has not been elucidated and was the purpose of this study. Our objective was to determine the role of IL-17A in airway inflammation and AR in a clinically relevant model of asthma exacerbation in which viral infection occurs during ongoing allergic airway inflammation. This is vital since therapeutics which inhibit IL-17A currently in clinical trials for asthma could have unintended effects on allergic airway inflammation.19
Methods
Mice
Pathogen-free 8–10-week-old female BALB/c mice were purchased from Charles River Laboratories. IL-17A KO mice on a BALB/c background were provided by Jay Kolls (University of Pittsburgh).
Allergic sensitisation/challenge and RSV infection of mice
Mice were categorised into four groups: mock, OVA, RSV and OVA/RSV. The protocol for OVA sensitisation/challenge and RSV infection is shown in figure 1 and has been previously described.5 ,7 RSV was provided by R Chanock (National Institutes of Health, Bethesda, Maryland, USA) and RSV stock was generated as previously described.20
Timeline of experimental protocol for ovalbumin (OVA)/respiratory syncytial virus (RSV) mice. OVA and OVA/RSV mice were sensitised with intraperitoneal (ip) injection of OVA complexed with Al(OH)3 (alum) on day –16. Mock and RSV mice were injected ip with alum on day –16. On days –2 through 5 OVA and OVA/RSV mice were challenged with 1% OVA (in phosphate buffered saline). On day 0, RSV and OVA/RSV mice were infected with RSV strain A2 and mock and OVA mice were challenged with uninfected cell culture supernatants.
Restimulation of mediastinal lymph nodes
Mediastinal lymph nodes (MLNs) were removed and homogenised from OVA, RSV or OVA/RSV mice on day 6. MLN cells were stimulated with no peptide (vehicle), OVA peptide 323–339 (1 μg/ml; Sigma-Aldrich), RSV M2 (127–135) peptide (10−7 M; Biosynthesis) or influenza peptide (Flu 147–155) (10−7 M) as a non-specific negative control (Biosynthesis). Cell culture supernatants were collected 24 h after stimulation.
Cytokine measurements
Cytokine levels were measured from whole lung homogenates or BAL fluid by ELISA (R & D Systems). Values below the limit of detection were assigned half the value of the lowest detectable standard.
Quantitative PCR
Quantitative PCR was conducted for IL-23p19 and GAPDH using SYBR green mix (BioRad). Primer sequences were as follows: IL-23p19 forward, 5′-TGGCTGTGCCTAGGAGTAGCA-3′, reverse, 5′-TTCATCCTCTTCTT CTCTTAGTAGATTCATA-3′; GAPDH, forward, 5′-GGCCCCTCTGGAAAGCTGTGG-3′, reverse, 5′-CCCGGCATCGAAGGTGGAAGA-3′.
AR measurements
Mice were anaesthetised with pentobarbital sodium (85 mg/kg), and a tracheostomy tube was placed and the internal jugular vein was cannulated. The mice were then placed in a plethysmography chamber and mechanically ventilated. Lung resistance was measured following administration of intravenous acetyl-β-methacholine (0–3700 μg/kg body weight; Sigma-Aldrich) as previously described.5 ,7
Statistical analyses
Data are presented as mean±SEM with data shown from one experiment. Each experiment was repeated three times to ensure the reproducibility of the data. In figures 2, 3 and ⇓5 the data were analysed by analysis of variance (ANOVA) followed by the Tukey post hoc test or a two-tailed t test with values being considered significant when p<0.05, and, in figure 4, the data were analysed by repeated measures ANOVA with the Bonferroni post hoc test. Values with p<0.05 were considered significant.
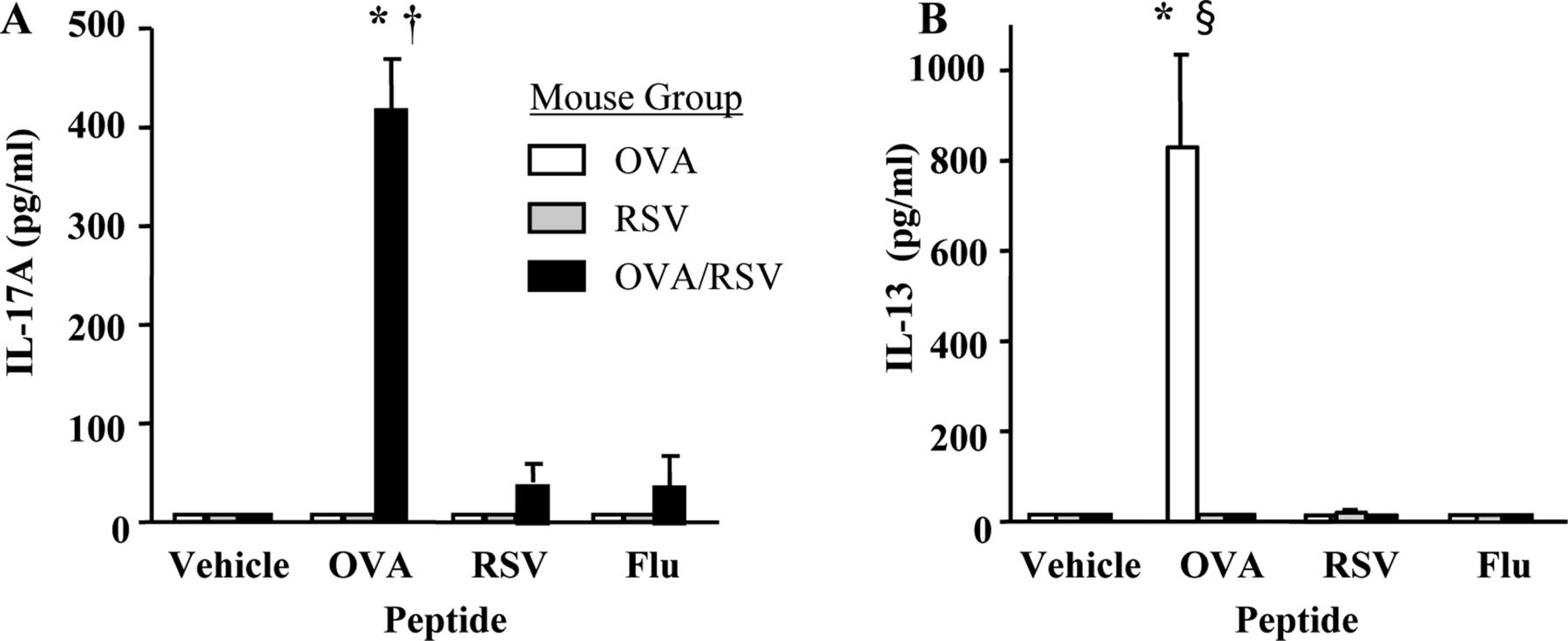
Interleukin 17 (IL-17) protein expression is increased in mediastinal lymph node (MLN) cells from ovalbumin (OVA), respiratory syncytial virus (RSV) or OVA/RSV mice restimulated with OVA peptide. MLN single cell suspensions from mice were restimulated with media alone (vehicle), OVA (323–339) peptide, RSV M2 (127–135) peptide or Flu (147–155) peptide for 24 h in vitro. (A, B) IL-17A and IL-13 protein expression in supernatants was examined by ELISA (n=4–5). *p<0.05 vs no peptide (vehicle); †p<0.05 vs OVA or RSV mouse groups; p<0.05 §vs RSV or OVA/RSV mouse groups (analysis of variance).
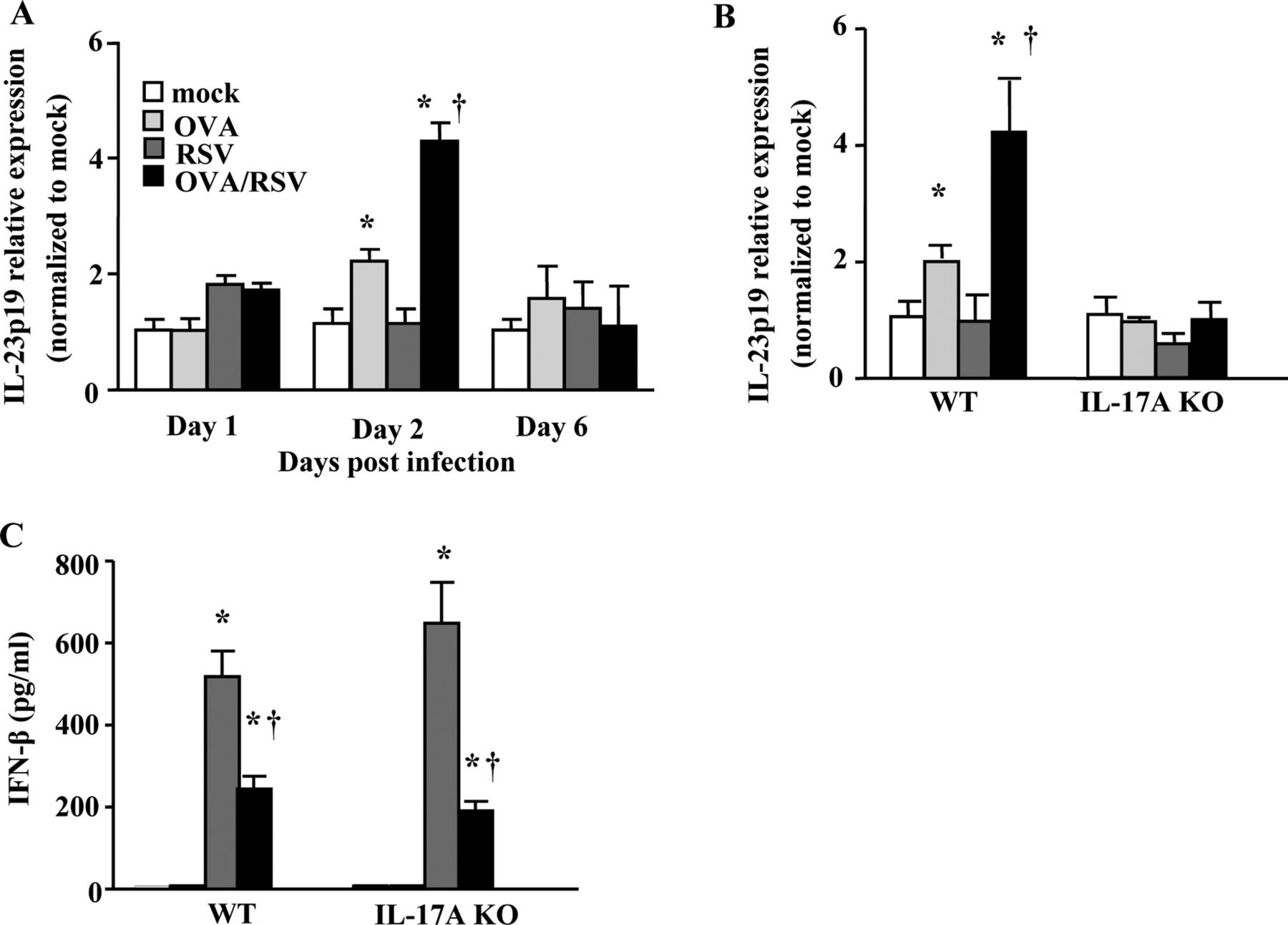
Ovalbumin (OVA)/respiratory syncytial virus (RSV) mice have increased lung interleukin (IL)-23p19 mRNA expression and decreased interferon (IFN)-β protein expression. (A) Lung IL-23p19 mRNA expression 1, 2 and 6 days after infection in wild-type (WT) mice. (B) Lung IL-23p19 mRNA expression 2 days after infection in WT and IL-17A knockout mice. In both A and B, samples were normalised to GAPDH and then mock animals. (C) IFN-β protein expression from whole lung homogenates 0.5 days after infection (n=5–6). *p<0.05 vs mock mice; †p<0.05 vs OVA mice (A–B) or RSV mice (C) (analysis of variance).

Interleukin (IL)-17A negatively regulates airway reactivity (AR) in ovalbumin (OVA)/respiratory syncytial virus (RSV) mice. Wild-type (WT) and IL-17A knockout mice were RSV-infected during ongoing allergic airway inflammation. On day 15, AR was measured by detecting changes in airway resistance in response to increasing concentrations of methacholine (n=7–15 mice). *p<0.05 vs WT mock mice, †p<0.05 vs WT OVA/RSV mice (repeated measures analysis of variance).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
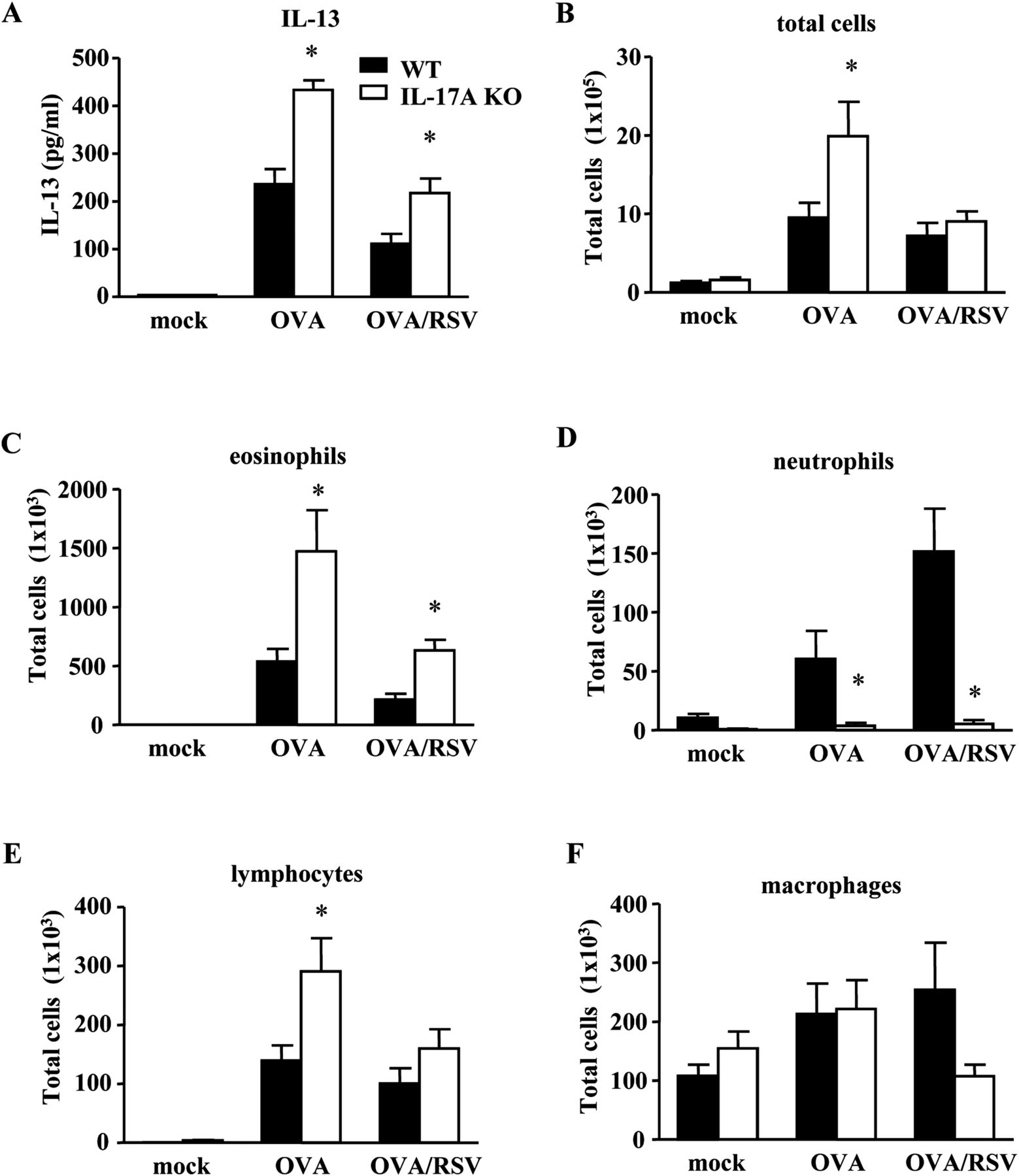
Interleukin (IL)-17A negatively regulates allergic airway inflammation in ovalbumin (OVA)/respiratory syncytial virus (RSV) mice. Wild-type (WT) and IL-17A knockout (KO) mice were RSV-infected during ongoing allergic airway inflammation. Bronchoalveolar lavage (BAL) fluid was harvested on day 2. (A) IL-13 protein expression from BAL fluid as measured by ELISA. (B–F) Infiltration of inflammatory cells in BAL fluid (n=5 mice). *p<0.05 vs WT mice for respective condition (analysis of variance).
Results
CD4 T cells express IL-17A protein in OVA/RSV mice
We previously found that lung IL-17A protein expression in lung homogenates peaked at day 6 and was significantly increased in OVA/RSV mice compared with OVA or RSV mice.8 Many cell types express IL-17A including γδ T cells, neutrophils, CD4 and CD8 T cells.21–23 We therefore wanted to determine the specific cell type responsible for IL-17A protein expression in OVA/RSV mice. We measured IL-17A protein expression by flow cytometry from the lungs of OVA/RSV mice on day 6. IL-17A expression was significantly increased in CD3CD4+ cells compared with CD3CD8, γδ TCR+, CD11c+ and Gr-1+ cells (see online supplementary figure S1A–F). Our data show that CD3CD4 T cells primarily produce IL-17A in OVA/RSV mice.
OVA peptide restimulation increases IL-17A protein expression in cells from MLNs
IL-17A protein expression was maximally increased in T cells from OVA/RSV mice, but we questioned whether OVA-specific or RSV-specific T cells were the source of IL-17A protein expression. OVA-specific T cells are present in the lung at the time of RSV infection, so we hypothesised that RSV infection increased the number of OVA-specific T cells expressing IL-17A. We harvested MLNs from OVA, RSV and OVA/RSV mice 6 days after infection and restimulated the cells with media alone (vehicle), OVA (323–339) peptide, RSV M2 (127–135) peptide or a non-specific control influenza peptide (Flu 147–155). IL-17A protein expression was examined in cell culture supernatants 24 h after stimulation. Only OVA/RSV MLN cells incubated with OVA peptide had significantly increased IL-17A protein expression compared with vehicle, RSV peptide or Flu peptide (figure 2A). Mice in the OVA or RSV groups had no detectable IL-17A protein expression with any peptide treatment. We also measured IL-13 expression from MLN cells. Only OVA MLN cells incubated with OVA peptide had a significant increase in IL-13 protein expression compared with vehicle, RSV peptide or Flu peptide (figure 2B). These data show that IL-17A protein expression is increased by OVA-specific T cells in OVA/RSV mice.
Lung IL-23p19 mRNA expression is increased by RSV infection during ongoing allergic airway inflammation
We next wanted to determine the mechanism for increased IL-17A in OVA/RSV mice. IL-23 is a cytokine required for Th17 cell sustainability and production of IL-17A,24–26 and therefore we examined lung mRNA relative expression of IL-23p19, a subunit of the IL-23 dimer, on days 1, 2 and 6 after infection in mock, RSV, OVA and OVA/RSV mice. We hypothesised that IL-23 is increased with RSV infection during ongoing allergic airway inflammation, leading to significantly increased IL-17A protein expression in OVA/RSV mice. Lung IL-23p19 relative mRNA expression was significantly increased on day 2 in OVA and OVA/RSV mice compared with mock and RSV mice (figure 3A). Lung IL-23p19 relative expression was significantly increased in OVA/RSV mice compared with OVA mice. RSV mice had no increase in lung IL-23p19 relative expression compared with mock mice. We also determined lung IL-23p19 mRNA expression in IL-17A KO mice on day 2 after infection (figure 3B). IL-17A KO mice had no change in lung IL-23p19 mRNA expression in OVA, RSV or OVA/RSV groups compared with the mock group. These data suggest that increased IL-23 expression in WT OVA/RSV mice leads to increased IL-17A protein expression and that IL-17A is required for IL-23p19 expression.
Pre-existing allergic airway inflammation decreases type I interferon response with RSV infection
Interferon β (IFN-β), a type I IFN, is significantly increased with RSV infection27 and negatively regulates IL-17A production.28 As lung IL-17A expression is increased in OVA/RSV mice compared with OVA mice, we hypothesised that OVA-induced allergic airway inflammation decreased lung IFN-β protein expression resulting from RSV infection in both WT and IL-17A KO mice. Lungs were harvested on day 0.5 (12 h) when IFN-β is maximally expressed (data not shown). In both WT and IL-17A KO mice, RSV-infected and OVA/RSV mice had significantly increased lung IFN-β protein compared with mock and OVA mice (figure 3C). OVA/RSV mice had a significant decrease in IFN-β protein expression compared with RSV mice in both WT and IL-17A KO mice. These data show that pre-existing allergic airway inflammation diminished IFN-β protein expression to viral infection. Furthermore, these data show that IL-17A does not regulate IFN-β protein expression.
IL-17A inhibits AR and airway inflammation in OVA/RSV mice
IL-17A was significantly increased only in the lungs of OVA/RSV mice,8 and OVA/RSV mice had increased AR.5 ,7 In addition, IL-17A has been associated with severe asthma in humans.9 ,10 We therefore hypothesised that IL-17A was responsible for the increased AR in OVA/RSV mice 15 days after infection and 9 days after the last exposure to aerosolised OVA. This is a time point at which AR in the OVA group was significantly diminished.5 We measured AR in mock, OVA and OVA/RSV groups in WT and IL-17A KO mice. To our surprise, IL-17A KO OVA/RSV mice had significantly increased AR compared with WT OVA/RSV mice (figure 4). In a separate experiment we analysed AR in WT RSV and IL-17A KO RSV mice. The magnitude of AR was lower in RSV mice compared with OVA/RSV mice, but IL-17A KO RSV mice had a significant increase in AR compared with WT RSV mice (see online supplementary figure S2). These data are in contrast to our hypothesis and suggest that IL-17A plays a regulatory role by inhibiting AR in the OVA/RSV model.
IL-17A inhibits IL-13 and infiltration of eosinophils in OVA/RSV mice
Since IL-17A negatively regulated AR in OVA/RSV mice, we examined IL-13 protein in BAL fluid on day 2, the point of maximal IL-13 protein expression in OVA/RSV mice.8 IL-17A KO OVA/RSV mice had significantly increased IL-13 protein expression compared with WT OVA/RSV mice and IL-17A KO OVA mice had significantly increased IL-13 compared with WT OVA mice (figure 5A).
To determine infiltration of inflammatory cells, we also examined inflammatory cells in BAL fluid on day 2 (figure 5B–F). IL-17A KO OVA/RSV mice had significantly increased eosinophils compared with WT OVA/RSV mice (figure 5C), and IL-17A KO OVA mice had significantly increased total inflammatory cells, eosinophils and lymphocytes compared with WT OVA mice (figure 5B, C and E). As IL-17A has been shown to be important in lung neutrophil recruitment, we hypothesised that IL-17A KO OVA/RSV mice have decreased airway neutrophils compared with WT OVA/RSV mice. IL-17A KO OVA/RSV mice had significantly decreased neutrophil infiltration compared with WT OVA/RSV mice (figure 5D). Taken together, these data suggest that IL-17A inhibits allergic airway inflammation by negatively regulating IL-13 protein expression and eosinophil and lymphocyte recruitment with RSV infection during ongoing allergic airway inflammation.
Discussion
Viral infections are a leading cause of asthma exacerbations in children and adults,1 ,2 and many of these patients have allergic airway inflammation.3 We have previously shown that IL-17A protein expression is increased in OVA/RSV mice.8 In this study we show that IL-17A is produced from OVA-specific CD3CD4 cells and not other cells known to produce IL-17A. We also found that OVA/RSV mice had increased IL-23p19 mRNA relative expression but decreased IFN-β protein expression in lung homogenates compared with OVA or RSV mice. Surprisingly, we found that IL-17A protected against AR and airway inflammation in the setting of viral infection during allergic airway inflammation, as IL-17A KO OVA/RSV mice had increased AR, IL-13 protein expression and eosinophil and lymphocyte infiltration compared with WT OVA/RSV mice.
RSV infections typically generate a Th1-mediated response with increased protein expression of IFN-γ, and allergic airway inflammation generates a Th2-mediated response with increased protein expression of IL-4, IL-5 and IL-13. However, RSV infection during ongoing allergic airway inflammation increased IL-17A protein expression and decreased IFN-γ and IL-13 protein expression.8 In our experiments, OVA-specific T cells were the cellular source of IL-17A, and there are several possible mechanisms by which RSV could increase OVA-specific CD4 T cell generation of IL-17A. First, RSV infection increased expression of lung IL-6,7 a cytokine that drives Th17 differentiation in mice.29 ,30 In addition, we found that RSV infection in the setting of allergic airway inflammation increased lung IL-23p19 mRNA expression, a cytokine important in the sustainability of the Th17 phenotype,29 while RSV infection by itself did not result in increased lung IL-23p19 expression. Therefore, increased IL-6 and IL-23 created the cytokine milieu that enhances Th17 cell differentiation.
We also found that allergic airway inflammation decreased IFN-β protein expression after RSV infection in OVA/RSV mice compared with RSV-infected mice alone. IFN-β, a type I IFN, negatively regulates IL-17A protein expression by increasing IL-27 protein expression from dendritic cells.28 ,31 Type I IFNs are also known to drive IFN-γ expression from Th1 cells. As allergic airway inflammation decreased RSV-induced lung IFN-β protein expression, this might explain the decreased lung IFN-γ expression in OVA/RSV mice compared with RSV mice. As IFN-γ is a negative regulator of IL-17A expression, the decreased IFN-γ in the OVA/RSV group might explain the increased lung IL-17A expression in OVA/RSV mice compared with OVA mice. Decreased expression of viral-induced type I IFNs and IFN-γ have been associated with asthma. RV-infected primary bronchial epithelial cells obtained from patients with asthma had a decreased type I IFN response compared with rhinovirus-infected bronchial epithelial cells from normal patients.32 Furthermore, IFN-γ was decreased in mononuclear cells of adult patients with asthma infected with RV.33 Together these data suggest that allergic airway inflammation decreases the IFN response to viral infections, consequently leading to increased IL-17A protein expression.
Other cells types such as CD8 T cells, γδ T cells and neutrophils secrete IL-17A,21–23 but we did not find IL-17A protein expression in these cells 6 days after infection. These cells may secrete IL-17A very shortly after RSV infection, but IL-17A was undetectable by ELISA in whole lung homogenates when examined at various time points after RSV infection so this is unlikely.8
IL-17A has been reported to be increased in the BAL fluid and bronchial biopsy tissue of patients with severe asthma.9 ,10 IL-17A protein expression, and not IL-13, was significantly increased in whole lung homogenates of OVA/RSV mice compared with OVA mice alone.8 We therefore hypothesised that IL-17A increased AR in OVA/RSV mice. In contrast to our hypothesis, IL-17A OVA/RSV KO mice had increased AR compared with WT OVA/RSV KO mice alone. No significant changes in AR were seen in IL-17A KO OVA mice compared with WT OVA mice 9 days after the last OVA aerosol challenge in the OVA experimental protocol. AR was increased in IL-17A KO RSV mice compared with WT RSV mice, but the magnitude of maximal AR was much smaller in the RSV group than in the OVA/RSV group for both WT and IL-17A KO mice. Th17 cells secrete other cytokines including IL-17F and IL-22, so we examined IL-17F and IL-22 protein expression in whole lung homogenates of WT and IL-17A KO OVA/RSV mice. No difference in lung IL-17F or IL-22 protein expression was detected in IL-17A KO OVA/RSV mice compared with WT OVA/RSV mice (data not shown), suggesting the increased AR in the IL-17A KO OVA/RSV mice was independent of IL-17F and IL-22 protein expression.
The role of IL-17A in mouse models of AR is conflicting in the literature. Adoptive transfer of OVA-specific Th17 cells into WT BALB/c mice or instillation of rmIL-17A into the airway increased AR in mice.16–18 Neutralisation of IL-17A prior to house dust mite exposure decreased AR and eosinophil infiltration into the airway,34 ,35 and IL-17A was enhanced by complement 3a signalling in this model.34 Furthermore, RSV A2 infection in mice deficient in complement 3a receptor or tachykinin receptor resulted in a significant decrease in IL-17A protein expression and AR.36 IL-17A also increased airway smooth muscle contraction.37 These results suggest that IL-17A may be responsible for increases in AR in allergen challenge and viral models. However, Schnyder-Candrian and colleagues reported that intranasal instillation of recombinant IL-17A into the airways of mice concurrent with OVA challenge in the OVA allergic airway model significantly decreased enhance respiratory pause (Penh) in response to methacholine, eosinophil infiltration and the chemokines CCL5, CCL11 and CCL17.13 Barlow and colleagues found that neutralising IL-17A increased airway eosinophilia and AR during a model of allergic airway inflammation.38 Further, IL-17A produced by γδ T cells protected against AR.15 We extend these findings by showing that IL-17A, produced by OVA-specific T cells, had a protective role on AR in a model of viral infection during allergic airway inflammation. Our findings are very important for understanding the pathogenesis of virally-induced asthma events.
IL-13 protein expression and eosinophil infiltration are significantly increased in the BAL fluid of IL-17A KO OVA/RSV mice compared with WT OVA/RSV mice. Our data provide a potential mechanism for increased AR in IL-17A KO OVA/RSV mice compared with WT OVA/RSV mice. However, future experiments in which IL-13 is neutralised with an antibody need to be conducted to definitively determine if increased IL-13 results in increased AR in IL-17A KO mice. These findings will be very important because clinical trials are currently planned to target IL-17A as a treatment for patients with inadequately controlled or steroid-resistant asthma.19 Our results suggest that neutralising IL-17A as a potential treatment for viral-induced asthma exacerbations may have unintended consequences of increased AR and airway inflammation. IL-17A is required for host immune responses to various strains of bacteria and fungi.12 ,39 Therefore, antagonising IL-17A or IL-17A signalling may also increase susceptibility to bacterial or fungal infections.
IL-17A has been associated with steroid-resistant asthma in an airway inflammation mouse model.16 IL-17A KO OVA/RSV mice have increased IL-13 protein expression and eosinophil recruitment to the lung compared with WT OVA/RSV mice, suggesting that IL-17A KO OVA/RSV mice could be developing a more Th2 steroid-responsive phenotype. It is plausible that anti-IL-17A therapeutics could potentially make patients with steroid-resistant asthma more responsive to corticosteroids, but experiments need to be conducted. Understanding the role of IL-17A in increased allergic airway inflammation and AR is vital for determining the impact of inhibiting or neutralising IL-17A as a treatment for patients with asthma.
Acknowledgments
We would like to thank William Lawson for use of his real-time PCR machine supported in part by a grant from National Institute of Health (1UL 1RR024975).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Funding National Institute of Health R01 HL 090664, R01 AI 070672, R01 AI 059108, GM 015431, R21 HL106446, U19AI095227, K12HD043483-08 and Veteran Affairs (1I01BX000624).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Ethics approval In caring for the animals, investigators adhered to the revised 1996 Guide for the Care and Use of Laboratory Animals prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council.
Linked Articles
- Chest clinic