Article Text

Abstract
Rationale Indoleamine 2,3-dioxygenase (IDO) induces generation of regulatory T cells but suppresses Th17 cells and therefore might attenuate neutrophilic inflammation. The role of IDO in neutrophilic airway diseases such as chronic obstructive pulmonary disease (COPD) remains unknown. We evaluated IDO activity and expression and interleukin (IL)-10 and IL-17A levels in sputum from patients with COPD.
Methods IDO activity and cytokine concentrations in sputum supernatants from patients with COPD of varying severity and in smoking and non-smoking control subjects were determined by high-performance liquid chromatography and ELISA, respectively.
Results Patients with COPD had reduced sputum IDO activity and expression and IL-10 levels, with increased IL-17A, IL-6 and CXCL8 concentrations and sputum neutrophils. These changes were significantly correlated with disease severity. IDO activity was decreased, but to a lesser extent, in normal smokers compared with non-smoking controls.
Conclusions Patients with COPD have a progressive reduction in IDO activity with reversal of the balance between IL-10 and IL-17A, resulting in chronic airway neutrophilic inflammation.
- COPD Mechanisms
Statistics from Altmetric.com
Key messages
What is the key question?
-
The key question is whether indoleamine 2, 3-dioxygenase (IDO) plays an important role in neutrophilic airway inflammation in COPD.
What is the bottom line?
-
In this study we demonstrated that IDO activity was progressively reduced in patients with COPD of increasing clinical severity which resulted in the induction of IL-17A and concomitant loss of IL-10 secretion.
Why read on?
-
In addition to in vivo data we also conducted in vitro study to confirm the effect of reduced IDO activity on the balance between IL-17A and IL-10 expression in monocyte-derived macrophages. The results may lead to a novel approach to treating inflammation in COPD by increasing IDO activity.
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic airway inflammatory disorder which involves an abnormal inflammatory response to noxious particles and gases. Cigarette smoking activates innate immune cells such as epithelial cells and macrophages.1 Activated dendritic cells induce adaptive immune cells including the two lineages of CD4 T helper (Th) cells (interferon (IFN)γ-secreting Th1 cells and interleukin (IL)-17-secreting Th17 cells), CD8 cytotoxic T cells (Tc1) and B cells.1 This leads to the formation of lymphoid follicles with chronic airway inflammation which is characterised by a predominance of CD8 T cells at all airway levels including the lung parenchyma.2 ,3 There is also an increase in CD68 cells (monocytes/macrophages) in the bronchial subepithelium and alveoli4 and an increased number of neutrophils in the airways and sputum. Previous studies have emphasised the role of several inflammatory cells in the pathogenesis of COPD—mainly alveolar macrophages, neutrophils and T lymphocytes.5 However, although proinflammatory CD4 T cell activation is involved in amplifying and perpetuating the chronic inflammatory state of stable COPD, the role of regulatory T (Treg) cells and CD4 Th17 cells remains uncertain.
There is an interplay between Treg and Th17 cells in regulating the development of autoimmunity and chronic inflammation.6 These lineages are both derived from naïve CD4 T cells, which undergo polarisation, and there is mutual suppression between these subtypes in vitro and presumably in vivo.7 The differentiation of Treg cells may be linked to the differentiation of Th17 cells depending on the overall polarising cytokine environment. The imbalance between Th17 and Treg cells may contribute to the chronic inflammatory response with reduced resolution of airway inflammation in COPD.
IL-17 is predominantly expressed by Th17 cells and has a potential proinflammatory role in COPD by stimulating the production of neutrophil chemotactic mediators such as CXCL1 (GRO-α), CXCL8 (IL-8) and IL-6 from airway epithelial cells.8 In addition, IL-17 is upregulated in the bronchial mucosa of patients with COPD.9 ,10 However, there are data that question the role of IL-17 in COPD as its expression is not well correlated with airway neutrophilia.9 ,10
Indoleamine 2,3-dioxygenase (IDO), the main inducible and rate-limiting tryptophan degrading enzyme, has recently been shown to act as an important T cell immunomodulator and inducer of tolerance.11 It is predominantly expressed in macrophages and dendritic cells.11 The IDO pathway has a potential role in the control of airway inflammation in asthma, and this may be mediated via the anti-inflammatory cytokine IL-1012 which is a potent activator of IDO.13 IDO inhibits T cell responses in various inflammatory disorders including autoimmune diseases, asthma, allograft rejection and viral infections. It regulates the balance of Th17 to Treg cells through the Treg differentiation inducible effects of kynurenine,14 ,15 thereby possibly supressing Th17-mediated inflammation. However, it is currently unknown whether COPD is associated with reduced IDO activity and how this affects the balance between Th17 and Treg cells. In addition to its anti-inflammatory effects, IDO could suppress oxidative stress by using superoxide anions as a substrate and a cofactor in its catalytic process.16 Furthermore, tryptophan catabolites act as potent radical scavengers.17 This led us to hypothesise that patients with increasing severity of COPD may have a progressive reduction in sputum IDO activity that may affect the balance between IL-10 and IL-17A. We therefore measured sputum IDO activity, IL-10 and IL-17A levels in patients with COPD of different severity compared with age-matched non-smoking and smoking controls.
We demonstrate here that patients with COPD have a reduction in IDO activity which is associated with increased IL-17A and decreased IL-10 concentrations and that this is correlated with disease severity. In addition, sputum IL-17A concentrations were positively correlated with IL-6 and CXCL8 concentrations at all COPD severity stages, whereas they were negatively associated with forced expiratory volume in 1 s/forced vital capacity (FEV1/FVC%).
Methods
Patients
Patients with COPD who were current or former smokers with a smoking history of ≥10 pack-years were recruited. We included male and female subjects aged 40–80 years with baseline post-bronchodilator FEV1/FVC < 0.7. COPD severity was graded according to the Global Initiative for Obstructive Lung Disease (GOLD).18 Patients were excluded if they had a history of asthma or a respiratory infection or a COPD exacerbation within 8 weeks prior to screening and sputum induction. Normal smoker controls had the same demographics and a similar smoking history but were free from significant diseases as determined by history and physical examination and had baseline post-bronchodilator FEV1 > 80% predicted normal value and FEV1/FVC > 0.7. Non-smoking controls were age-matched to normal smokers but with no smoking history. The study was approved by the ethics review committees of Siriraj Hospital and all subjects gave written informed consent.
Study design
This study was aimed at investigating the levels of sputum IDO activity in patients with COPD of different severity. All patients entering the run-in phase stopped inhaled corticosteroids (ICS), theophylline and long-acting bronchodilators (LABA) and received inhaled short-acting β2 agonists used as rescue medication for 2 weeks prior to sputum induction. At the end of the run-in period, patients were eligible for the study if they had complied with the run-in treatment and had no COPD exacerbations. Seven patients with GOLD stage III COPD dropped out in the run-in period when ICS, theophylline and LABA or long-acting muscarinic antagonists had been withdrawn.
Statistical analysis
Data are presented as means±SEM. Demographic data were described by number and percentage of patients for categorical variables. Comparisons of IDO activity between patients with COPD who were current or ex-smokers and between cigarette smoke extract (CSE)-untreated and CSE-treated macrophages were performed by the Mann–Whitney U test. The rs correlation coefficient was determined for the correlation of the kynurenine/tryptophan (Kyn/Trp) ratio, IL-10 and IL-17A with FEV1/FVC% using the Spearman rank correlation test. Multiple linear regression analyses were performed for independent variables (medication, number of pack-years). The median was calculated if the distribution of the variables was not normal. Statistical analysis for multiple comparisons was performed using one-way ANOVA and the Welch test with the Dunnett T3 correction19 for equal variances not assumed or Bonferroni corrections for equal variances assumed. All statistical tests were two-sided, and significance was accepted at the level of 95% and p<0.05 using PASW statistics 18 (SPSS, IBM, Somers, New York, USA).
Additional details of the study method are provided in the online supplement.
Results
Subject characteristics
We studied 63 subjects: 44 with COPD (10 GOLD stage I, 20 GOLD stage II, 14 GOLD stage III), 12 smoking controls without COPD (9 current smokers) and 7 never smokers with normal lung function (table 1). The percentage of sputum neutrophils increased with GOLD grades.
Demographic data and clinical characteristics of study subjects
IDO expression and activity
IDO expression and bioactivity (expressed as the Kyn/Trp ratio) measured in sputum samples were both decreased in patients with COPD compared with non-smoking controls, and this was correlated with disease severity (figure 1A,B). These differences reached significance in samples from smoking normal controls (IDO immunoreactive cells 43.2±1.5%, p<0.001; Kyn/Trp ratio 1.36±0.18, p<0.001), GOLD stage I COPD (IDO immunopositive cells 29.5±3.4%, p<0.001; Kyn/Trp ratio 0.58±0.07, p<0.001), GOLD stage II COPD (IDO immunopositive cells 15.6±1.3%, p<0.001; Kyn/Trp ratio 0.17±0.03, p<0.001) and GOLD stage III COPD (IDO immunopositive cells 5.8±0.5%, p<0.001; Kyn/Trp ratio 0.048±0.009, p<0.001) compared with those from non-smoking controls (IDO immunopositive cells 76.28±2.7%; Kyn/Trp ratio 2.55±0.1). There was a graded reduction in IDO expression and bioactivity in patients with increasing clinical severity of COPD: for IDO expression (smoking controls vs GOLD stage I, p=0.025; GOLD stage I vs GOLD stage II, p=0.021; GOLD stage II vs GOLD stage III, p<0.001); for IDO activity (smoking controls vs GOLD stage I, p=0.012; GOLD stage I vs GOLD stage II, p=0.004; GOLD stage II vs GOLD stage III, p=0.011). Concentrations of kynurenine were also significantly reduced in sputum samples from patients with increasing severity of COPD compared with samples from non-smoking controls (figure 1C). The differences were significant between samples from non-smoking controls (69.6±4.5 μM) and those from normal smokers (19.7±1.7 μM, p<0.001), GOLD stage I COPD (10.4±0.78 μM, p<0.001), GOLD stage II COPD (6.3±0.72 μM, p<0.001) or GOLD stage III COPD (2.9±0.26 μM, p<0.001). The graded reduction in kynurenine levels also corresponded with the increasing severity of COPD (smoking controls vs GOLD stage I, p=0.002; GOLD stage I vs GOLD stage II, p=0.009; and GOLD stage II vs GOLD stage III, p=0.001). In addition, IDO activity was significantly correlated with the degree of airflow obstruction (FEV1/FVC %) (rs=0.54, p=0.0001; figure 1D) and the number of pack-years (coefficient of variance 0.003, p=0.022). There was no difference in IDO activity between patients with COPD who were treated with ICS/LABA (coefficient of variance 0.085, p=0.51). However, there was no significant difference in IDO activity between patients with COPD who were current smokers or ex-smokers (current smokers 0.31±0.09 vs ex-smokers 0.22±0.04, p=0.4).

Comparison of (A) sputum indoleamine 2,3-dioxygenase (IDO) expression, (B) IDO activity and (C) kynurenine concentrations in patients with chronic obstructive pulmonary disease of different Global Initiative for Obstructive Lung Disease (GOLD) stages, normal smokers and non-smokers. (D) A positive correlation is shown between IDO activity and forced expiratory volume in 1 s/forced vital capacity (FEV1/FVC%). Results expressed as means±SEM. *p<0.05 vs non-smokers; **p<0.001 vs non-smokers. Kyn/Trp ratio, kynurenine/tryptophan ratio.
IL-10
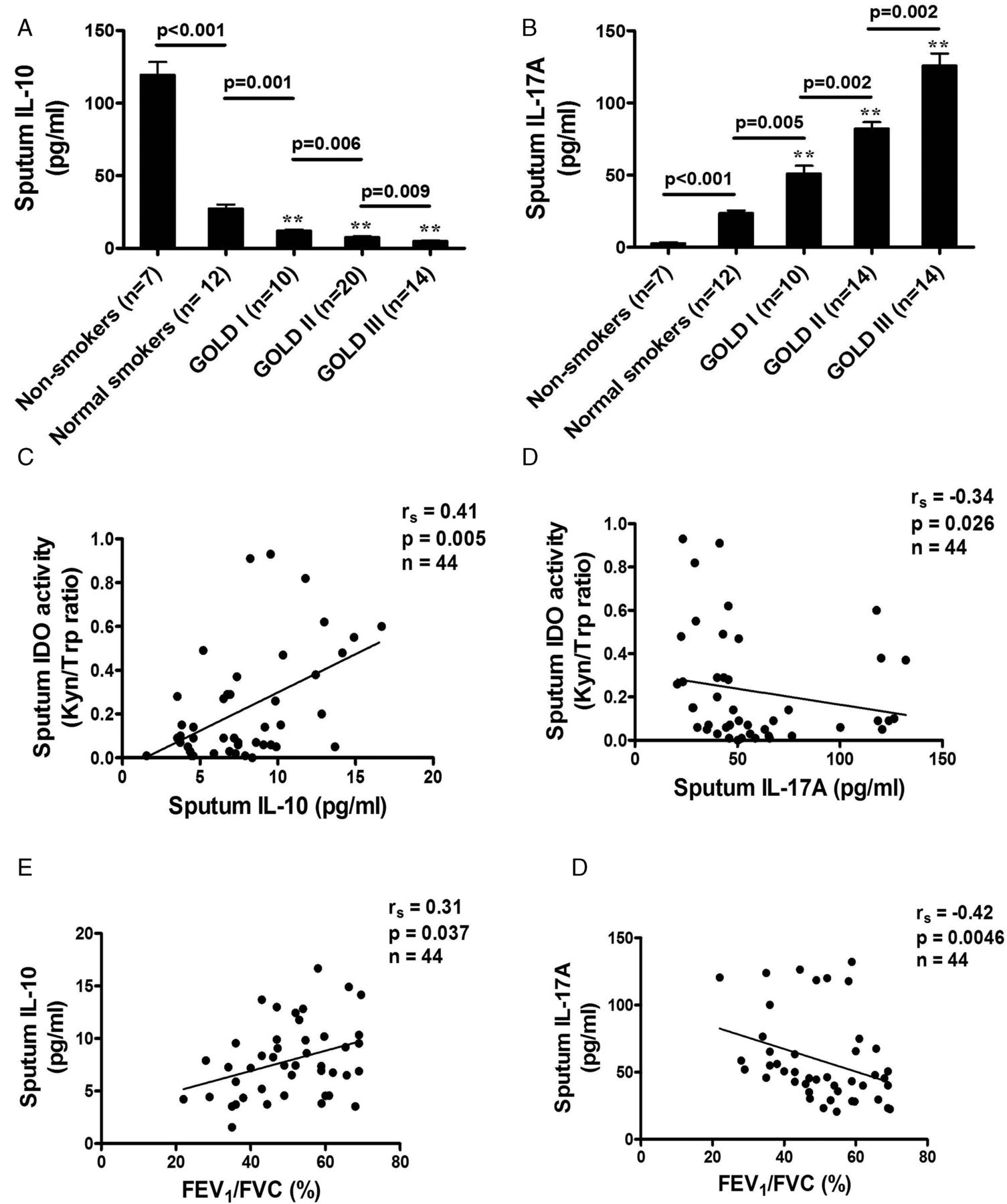
Concentrations of IL-10 were lower in sputum samples from patients with more severe COPD than in samples from non-smoking controls. A significant reduction was found in sputum samples from smoking control subjects (27.5±2.7 pg/ml) compared with samples from non-smoking controls (119.5±8.9 pg/ml, p<0.001) and from patients with GOLD stage I COPD (12.1±0.84 pg/ml, p<0.001; figure 2A). A further significant reduction was detected in samples from patients with GOLD stage III disease (5.0±0.49 pg/ml) compared with samples from non-smoking controls (p<0.001) and those with GOLD stage II disease (7.83±0.59 pg/ml, p<0.001). Patients with COPD of greater clinical severity exhibited a greater reduction in sputum IL-10 levels (smoking controls vs GOLD stage I, p=0.001; GOLD stage I vs GOLD stage II, p=0.006; GOLD stage II vs GOLD stage III, p=0.009). In addition, sputum IL-10 levels were significantly correlated with IDO activity (rs=0.41, p=0.005) and the degree of airflow obstruction (rs=0.31, p=0.037; figure 2C,E).
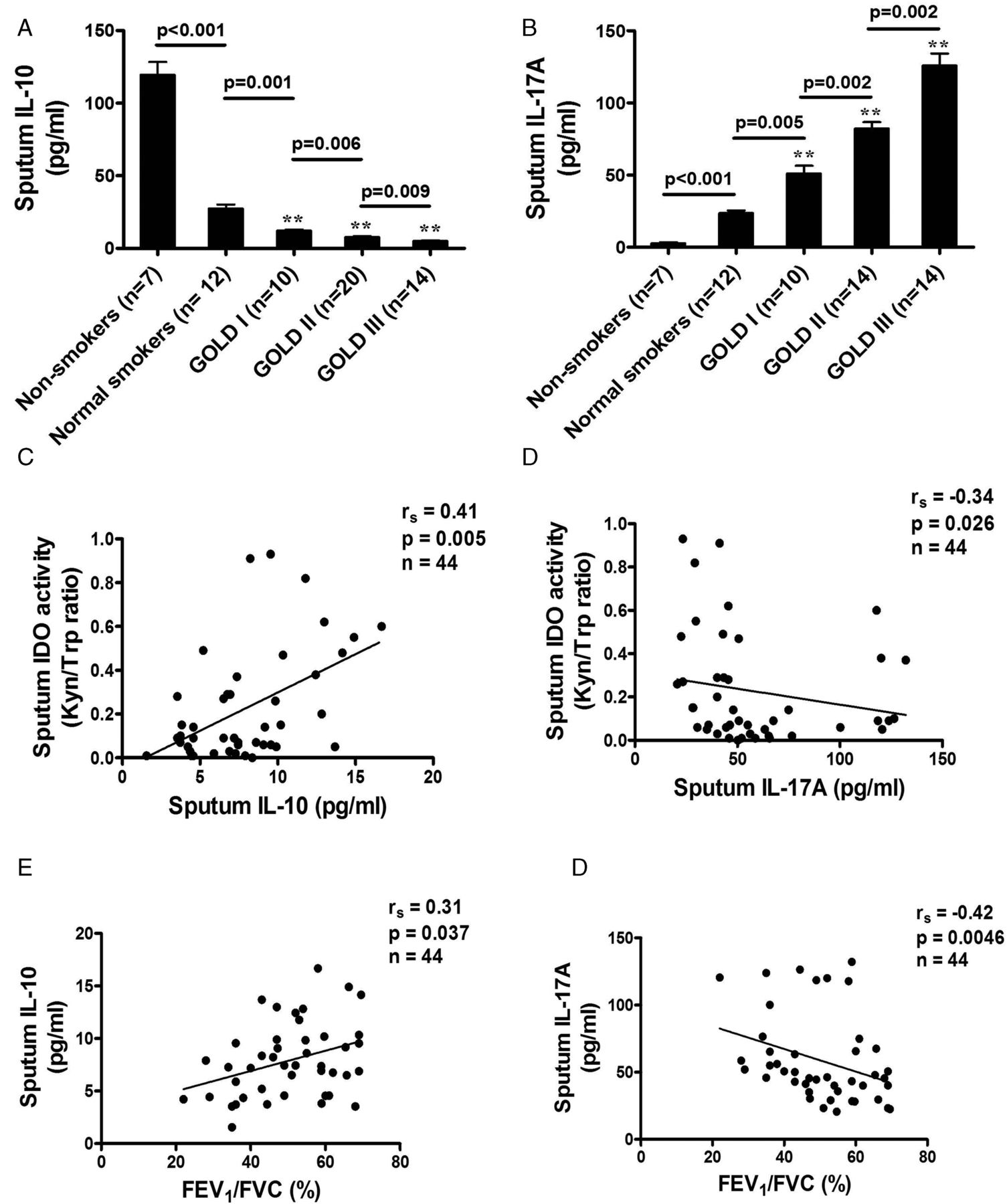
Comparison of sputum concentrations of (A) interleukin (IL)-10 and (B) IL-17A in patients with chronic obstructive pulmonary disease of different Global Initiative for Obstructive Lung Disease (GOLD) stages, normal smokers and non-smoking controls. (C, D) Relationship between sputum indoleamine 2,3-dioxygenase (IDO) and IL-10 and IL-17A in patients of all GOLD stages showing the association of lower sputum IDO concentrations in patients with lower IL-10 levels and in patients with higher IL-17A levels. (E, F) Relationship between forced expiratory volume in 1 s/forced vital capacity (FEV1/FVC%) and IL-10 and IL-17A in patients of all GOLD stages showing an association of lower FEV1/FVC% in patients with lower IL-10 levels and in patients with higher IL-17A levels. Results expressed as means±SEM. **p<0.001 vs non-smokers.
IL-17A
Patients with COPD of greater severity had an increase in sputum IL-17A concentrations (smoking controls vs GOLD stage I, p=0.005; GOLD stage I vs GOLD stage II, p=0.002; GOLD stage II vs GOLD stage III, p=0.002). A significant increase was found in sputum samples from smoking controls (23.6±1.8 pg/ml) compared with samples from non-smoking controls (2.6±0.7 pg/ml, p<0.001) and patients with GOLD stage I COPD (51.1±5.4 pg/ml, p<0.001; figure 2B). A further significant increase was detected in samples from patients with GOLD stage II COPD (82.2±4.4 pg/ml) compared with samples from non-smoking controls (p<0.001) and from patients with GOLD stage III COPD (125.9±8.3 pg/ml, p<0.001). Sputum IL-17A levels in COPD patients were inversely correlated with IDO activity (rs=−0.34, p=0.026; figure 2D) and the degree of airflow obstruction (rs=−0.42, p=0.0046; figure 2F). The upregulation of IL-17A levels was correlated with increased sputum IL-6 concentrations (rs=0.47, p=0.0011) and CXCL8 concentrations (rs=0.36, p=0.016) in patients with COPD of all severity stages (figure 3A,B) but not with percentage sputum neutrophils (rs=0.27, p=0.07; figure 3C), whereas increased CXCL8 concentrations were strongly correlated with percentage sputum neutrophils (rs=0.66, p<0.001; figure 3D).
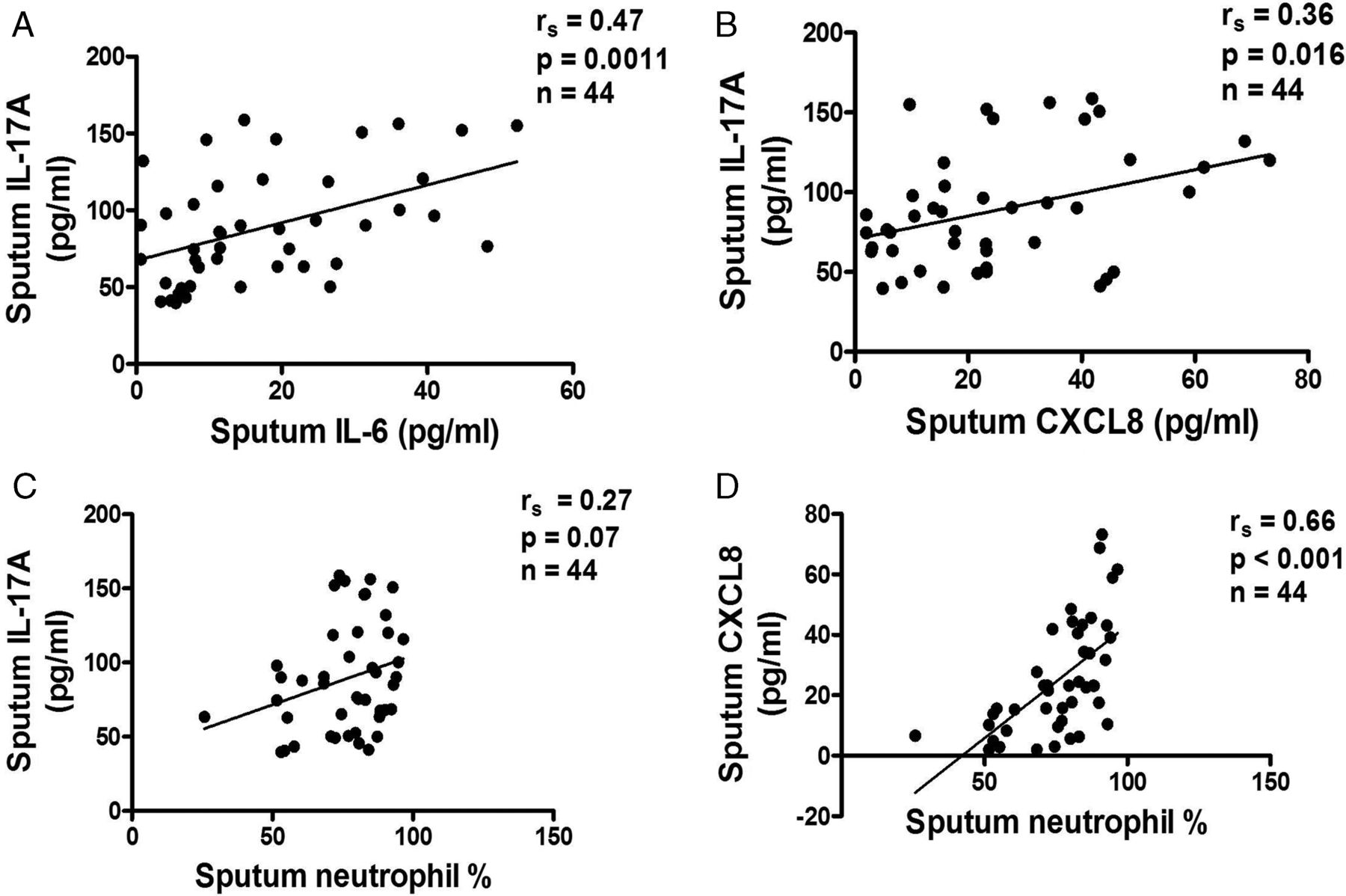
3 Relationship between sputum interleukin (IL)-17A levels and (A) IL-6, (B) CXCL8, (C) sputum neutrophil percentage in patients with chronic obstructive pulmonary disease of all Global Initiative for Obstructive Lung Disease (GOLD) stages, illustrating a positive correlation of IL-17A in patients with IL-6 and with CXCL8 but no correlation with sputum neutrophil percentage. (D) A positive correlation is shown between CXCL8 levels and sputum neutrophil percentage in patients with all GOLD stages.
In vitro experiments
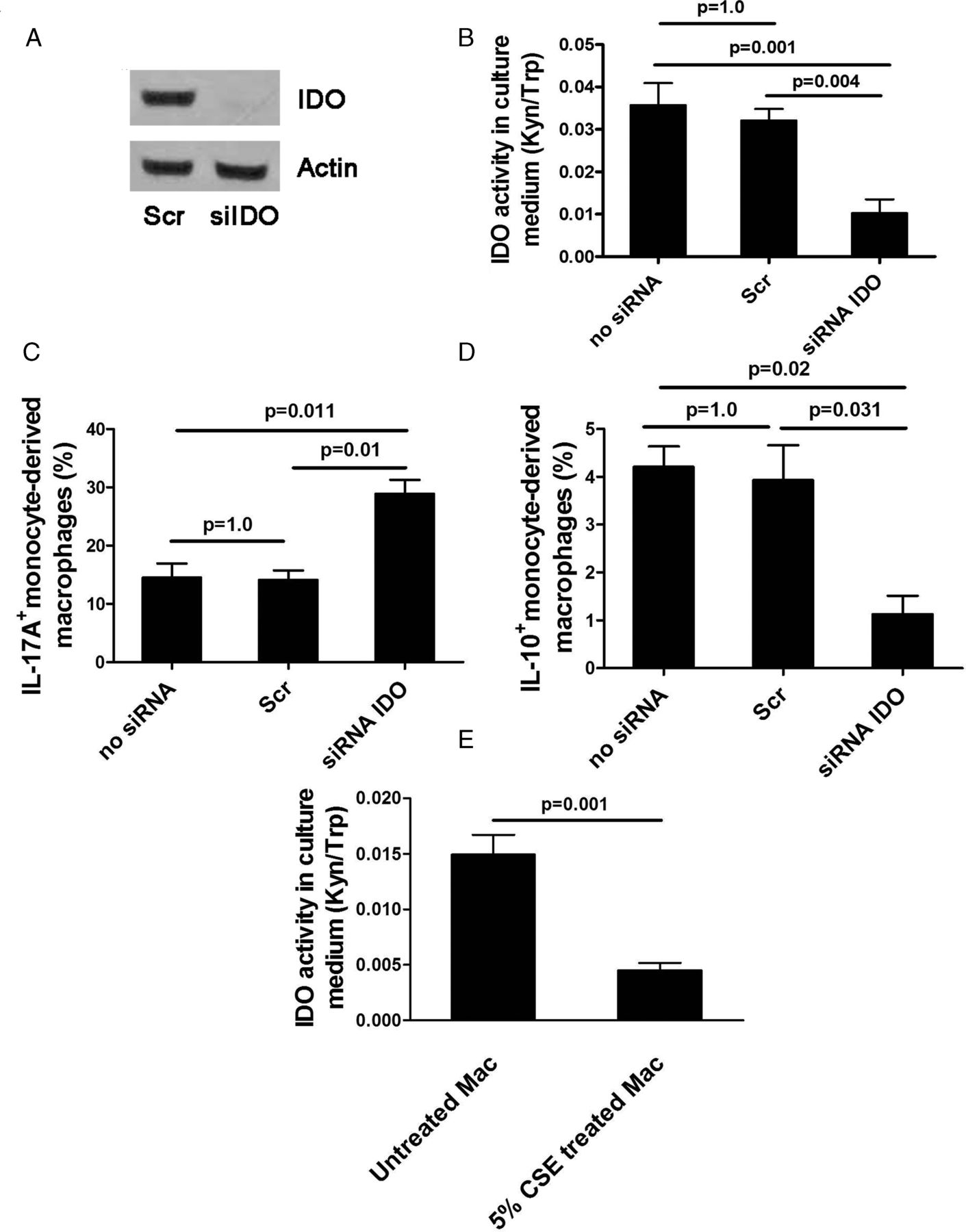
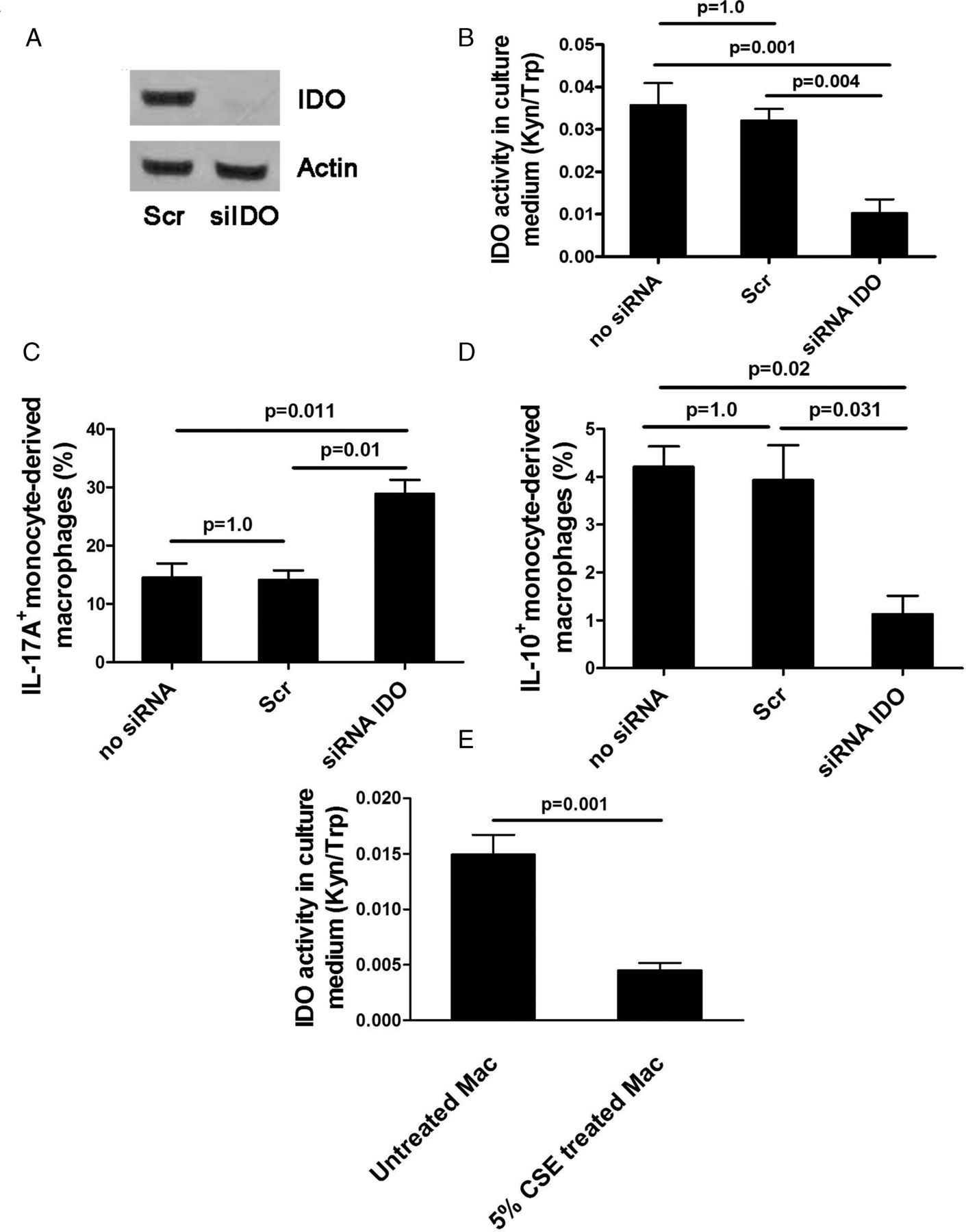
We conducted an in vitro study to investigate the effect of reduced IDO activity on the balance between IL-17A and IL-10 expression in monocyte-derived macrophages by knockdown of IDO with siRNA. Treatment of macrophages with siRNA IDO resulted in IDO knockdown both its mRNA and protein expression and bioactivity (Kyn/Trp ratio 0.01±0.003, p=0.004) compared with scrambled siRNA (0.03±0.002; figure 4A,B), with 77% reduction in IDO mRNA expression. IDO knockdown significantly tipped the balance of the expression of these two cytokines towards IL-17A whereas control siRNAs had no effect (IL-17A: 28.8±2.4% for siRNA IDO vs 14.1±1.6% for scrambled siRNA, p=0.01; IL-10: 1.1±0.38% vs 3.9±0.7%, p=0.031; figure 4C,D). In addition, CSE significantly reduced IDO activity in monocyte-derived macrophages (CSE-treated macrophages 0.004±0.0006 vs CSE-untreated macrophages 0.014±0.001, p=0.001; figure 4E). These in vitro results complemented our in vivo finding that cigarette smoke contributed to the loss of IDO activity which resulted in IL-17A/IL-10 imbalance.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of siRNA knockdown of indoleamine 2,3-dioxygenase (IDO) in macrophages treated with siIDO compared with scramble (Scr) siRNA. (A) IDO protein expression, (B) IDO activity, (C) interleukin (IL)-17A expression, (D) IL-10 expression, (E) IDO activity in monocyte-derived macrophages exposed to cigarette smoke extract (CSE). Mean±SEM values of three independent experiments are shown. Kyn/Trp ratio, kynurenine/tryptophan ratio.
Discussion
In this study we demonstrate for the first time that IDO activity and expression is reduced in the sputum of patients with COPD and that this is related to increasing clinical severity. In addition, patients with COPD of increasing clinical severity had progressive upregulation of IL-17A and reduction in IL-10 concentrations. Increased sputum IL-17A concentrations were in turn positively correlated with IL-6 and IL-8 concentrations and with sputum neutrophils.
IDO mediates T cell suppressive and antioxidant effects. It inhibits T cell-mediated responses in several inflammatory disorders, all of which are associated with either neutrophilic or eosinophilic inflammation. It uses superoxide anions as a substrate and a cofactor in its catalytic process.16 On consumption of superoxide anions, IDO initiates the formation of tryptophan catabolites which act as potent radical scavengers.17 Because of its ability to convert a pro-oxidant into antioxidants, IDO modulates oxidative stress.17 IDO suppresses the formation of intracellular reactive oxygen species in acute lung allograft injury and enhances the resistance of lung cells to oxidative stress.20 Although no data exist on the expression of IDO in COPD, the presence of oxidative stress in patients with COPD,21 at least in part due to increasingly impaired antioxidant enzymes,22 may imply a reduction in IDO activity. Our study showed that IDO activity was indeed reduced in normal smokers who did not have airflow obstruction compared with healthy non-smokers, and progressively reduced with increasing severity of COPD. This reduction in IDO activity was, at least in part, due to decreased IDO expression in sputum macrophages.
Kynurenine, the major metabolite of IDO activity, may tip the balance of IL-10-producing Treg cells to IL-17-producing Th17 cells.14 Our data confirm that this occurs in COPD patients and that the reduction of IDO activity is linked to the change in balance of IL-10/IL-17 towards IL-17A in induced sputum. A greater imbalance of IL-10/IL-17A levels was found if the clinical severity of COPD was greater, consistent with a previous study showing that the degree of IL-17 upregulation in the bronchial mucosa of patients with stable COPD was correlated with disease severity.9 In addition, patients with more severe COPD had a significantly greater reduction in kynurenine concentrations. These data suggest that the IL-10/IL-17A balance may be regulated by IDO through the effects of kynurenine.
IL-17A was upregulated in the bronchial mucosa of patients with COPD9 ,10 and may contribute to the regulation of chronic inflammation in COPD.9 IL-17 induced the release of CXCL1, CXCL8 and granulocyte–macrophage colony-stimulating factor from airway epithelial cells and smooth muscle cells, thereby orchestrating neutrophilic inflammation.8 ,23 ,24 Mice exposed to cigarette smoke show upregulation of IL-17 production and increased airway neutrophilia,25 and the inhibition of IL-17 by a specific neutralising antibody attenuates neutrophilic airway inflammation.26 However, the present study is consistent with previous studies9 ,10 that found no correlation between sputum IL-17A levels with neutrophilic airway inflammation, whereas sputum IL-17A concentrations were negatively associated with airflow obstruction (FEV1/FVC%) in patients with COPD. Sputum IL-17A levels were also associated with IL-6 and CXCL8 concentrations which correlated with the percentage of sputum neutrophils. These data suggest that IL-17A-induced neutrophilic airway inflammation in COPD is mediated, at least in part, through IL-6 and CXCL8. However, we cannot exclude the possibility that CXCL8 might be regulated independently from IL-17A as several stimulants are involved in CXCL8 production through the activation of Toll-like receptor (TLR) 9 on neutrophils or via the activation of airway epithelial cells by human rhinovirus and cigarette smoke.27–29
The reduction in sputum kynurenine and IL-10 appears to be smoking-dependent. In patients with COPD, although the regulation of IL-10 production is complex, this, at least in part, is associated with smoking status and is dependent on cell type. Cigarette smoking may promote IL-10 release from the airway that is primarily defective in patients with COPD.30 Cigarette smoke exposure could induce IL-10 production through the upregulation and activation of TLR4 and TLR9 on lung CD8 T cells from patients with COPD.31 The number of mucosal mature dendritic cells that produce IL-10 was decreased in healthy smokers and current smokers with COPD,32 ,33 and this might reduce IL-10 concentrations in the airways. In addition, kynurenine induces the development of tolerogenic dendritic cells14 and the differentiation of CD4 T cells towards Treg cells15 through the interaction with aryl hydrocarbon receptors that results in the induction of IL-10 and tryptophan catabolism. This suggests that the IL-10 reduction in smokers, which was not seen in patients with COPD, may be due to the fact that the majority of them in this study were ex-smokers.
In contrast, IL-17A and IDO activity were COPD-dependent. The IDO molecule contains heme, which is essential for its enzymatic activity. The activation of IDO requires the single electron reduction of ferric to ferrous iron that promotes the binding of IDO to L-tryptophan. Intracellular reducing cofactors including superoxide anion radical are required for IDO heme iron reduction. Cigarette smoke-induced oxidative stress increases superoxide anion radical levels34 and particulate matter in cigarette smoke causes accumulation of iron,35 both of which might affect IDO activity. This was consistent with our in vitro study which showed that CSE significantly suppressed IDO activity in macrophages. Although the number of pack-years was positively correlated with IDO activity, there was no difference in IDO activity between patients with COPD who were current smokers and patients with COPD who were ex-smokers. This may explain the higher IDO activity in healthy subjects than in patients with COPD, most of whom were ex-smokers. The mechanisms of IDO suppression in patients with COPD could involve other alternative pathways that warrant further investigation.
Statin treatment upregulates IDO activity in patients with asthma36 and tips the balance between Treg and Th17-derived cytokines towards IL-10, with downregulation of IL-17.37 In addition, the use of statins has been associated with a reduced number of exacerbations and the rate of FEV1 decline in patients with COPD.38 ,39 Statins may therefore be beneficial in suppressing airway inflammation in patients with COPD, and this requires future investigation in controlled trials.
In summary, our data provide evidence that IDO activity is progressively reduced in patients with COPD of increasing clinical severity which results in the induction of IL-17A and concomitant loss of IL-10 secretion. This suggests that inflammation in patients with COPD may be amplified, and that this may be a factor determining disease severity and long-term progression of the disease. It also raises the possibility that increasing IDO activity may be a novel approach to treating inflammation in COPD.
Acknowledgments
We thank Tasaneeya Suthamsamai, Jirawat Assawapoom and Kanokwan Ratanasaenglert for technical assistance and Valla Wamanuttajinda for assistance with in vitro experiments. We also thank the normal volunteers for their participation in the study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors Study concept: KM. Study experiments: KM, KS. Data analysis: KM, KS, AW. Manuscript preparation: KM, PJB.
-
Funding The study was supported by the National Science and Technology Development Agency (NSTDA) (BT-B-01-MG-14-5114).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The Ethics Review Committees of Siriraj Hospital.
-
Provenance and peer review Not commissioned; externally peer reviewed.