Article Text

Abstract
Background Overexpression of the transforming growth factor β family signalling molecule smad2 in the airway epithelium provokes enhanced allergen-induced airway remodelling in mice, concomitant with elevated levels of interleukin (IL)-25.
Objective We investigated whether IL-25 plays an active role in driving this airway remodelling.
Methods Anti-IL-25 antibody was given to mice exposed to either inhaled house dust mite (HDM) alone, or in conjunction with an adenoviral smad2 vector which promotes an enhanced remodelling phenotype.
Results Blocking IL-25 in allergen-exposed mice resulted in a moderate reduction in pulmonary eosinophilia and levels of T helper type 2 associated cytokines, IL-5 and IL-13. In addition, IL-25 neutralisation abrogated peribronchial collagen deposition, airway smooth muscle hyperplasia and airway hyperreactivity in control mice exposed to HDM and smad2-overexpressing mice. IL-25 was shown to act directly on human fibroblasts to induce collagen secretion. Recruitment of endothelial progenitor cells to the lung and subsequent neovascularisation was also IL-25 dependent, demonstrating a direct role for IL-25 during angiogenesis in vivo. Moreover, the secretion of innate epithelial derived cytokines IL-33 and thymic stromal lymphopoietin (TSLP) was completely ablated.
Conclusions In addition to modulating acute inflammation, we now demonstrate a role for IL-25 in orchestrating airway remodelling. IL-25 also drives IL-33 and TSLP production in the lung. These data delineate a wider role for IL-25 in mediating structural changes to the lung following allergen exposure and implicate IL-25 as a novel therapeutic target for the treatment of airway remodelling in asthma.
- Airway Epithelium
- Allergic lung disease
- Asthma
- Asthma Mechanisms
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Key messages
What is the key question?
-
Does interleukin (IL)-25 play a functional role in airway remodelling in allergic airways disease?
What is the bottom line?
-
IL-25 mediates pulmonary collagen deposition, neovascularisation, peribronchial smooth muscle hyperplasia and airway hyperreactivity following allergen exposure.
Why read on?
-
IL-25 is implicated as a novel therapeutic target for the treatment of airway remodelling in asthma.
Introduction
Asthma affects 300 million people worldwide and while current therapeutics may control eosinophilic and T helper type 2 (Th2)-driven inflammation, treatments for the associated airway remodelling which encompasses goblet cell hyperplasia, fibrosis, increased airway smooth muscle (ASM) mass and neovascularisation are lacking. Therefore, identification of molecules underlying airway remodelling is a priority. Interleukin (IL)-25 was initially described as an amplifier of Th2 immune responses by inducing Th2 cell cytokines such as IL-13, IL-4 and IL-5.1 ,2 Direct delivery of IL-25 to the lungs results in airway hyperreactivity (AHR) and eosinophilic pulmonary inflammation that develops via IL-13 and the Stat-6 pathway.3 Conversely, blockade of IL-25 during acute ovalbumin-driven allergic airways disease abrogates AHR.4 A wider role for IL-25 in development of effector T-cell responses has been outlined with the discovery that IL-25 can potentiate secretion of IL-9 from Th9 cells5 and, in combination with IL-33, can promote the development of type 2 innate lymphoid cells which are critical in the early initiation of Th2 responses.6 ,7
Perturbations of the airway epithelium are capable of driving structural changes linked to airway remodelling.8 We previously used an adenovirus encoding the transforming growth factor (TGF)-β/activin A signalling molecule smad2 to overexpress this protein in airway epithelium before exposure to inhaled house dust mite (HDM). This resulted in an enhanced remodelling phenotype compared with allergen exposure alone and identified a novel role for activin A in mediating AHR and remodelling. IL-25 expression is also triggered in the HDM-exposed mice overexpressing smad2 and neutralisation of activin A completely abolished the elevated IL-25, implicating a potential role for IL-25 in remodelling responses.
Support for this hypothesis comes from in vitro studies that suggest that IL-25 may also contribute to angiogenesis.9 Biopsies of patients with asthma show elevated levels of IL-25 and IL-17RB (its receptor) transcripts compared with control subjects without asthma/atopy10 ,11 and elevated expression of IL-25 is inversely correlated with forced expiratory volume in 1 s in patients with asthma.9 However, a potential functional role for IL-25 in modulating remodelling has not yet been investigated.
The majority of data accumulated so far for IL-25 focus on the early initiation of Th2 responses, therefore we investigated whether IL-25 contributes to aspects of pathology associated with prolonged allergen exposure. Inhaled HDM induces phenotypic changes in mice characteristic of human asthma and we determined the effects of blocking endogenously produced IL-25 on parameters of remodelling in control mice and those overexpressing smad2 which have an enhanced remodelling phenotype. Our data suggest that in addition to its proven role in the initiation of effector type 2 responses, IL-25 also drives the airway remodelling responses that typify chronic disease.
Materials and methods
Animals
Female BALB/c mice (Charles River), 6–8 weeks old, received 15 μg HDM extract (Dermatophagoides pteronyssinus in phosphate-buffered saline (PBS)) (Greer laboratories, Lenoir, North Carolina, USA) or 15 μl PBS intranasally 3 days a week for up to 3 weeks. Some groups received a first-generation replication-deficient adenovirus serotype 5 containing murine smad2 cDNA (AdSmad2) (2×109 viral particles in 25 μl PBS) or an empty vector lacking a transgene (AdC) 2 days prior to commencing instillation of either HDM or PBS. In addition, mice received either 500 μg of neutralising antibody to murine IL-254 or control IgG (R&D Systems, Abingdon, UK) via intraperitoneal injection 2 h prior to intranasal challenge with PBS or HDM. All experiments were performed in accordance with UK Home Office guidelines.
Measurement of AHR
Airway responsiveness was determined by direct measurements of resistance and compliance in anaesthetised and tracheostomised mice following 3 weeks of HDM challenge.8
Sample preparation
Serum, bronchoalveolar lavage (BAL) and lung cells were collected.10–12 Differential cell counts were performed on Wright-Giemsa-stained cytospins. Paraffin-embedded sections (4 μm) were stained with H&E, periodic acid-Schiff (PAS) and Sirius red. All scoring and measurements were performed blinded by the same observer on medium airways measuring between 150 and 250 μm in diameter.8 Paraffin sections were stained with rabbit anti-mouse proliferating cell nuclear antigen (PCNA) (Abcam, Cambridge, UK), α-smooth muscle actin (α-SMA) (Abcam), von Willebrand factor (vWF) (Dako, Aachen, Germany), IL-25 (Millipore, Billerica, Massachusetts, USA) or IL17RB (Bioss, Woburn, Massachusetts, USA).13–15
Quantification of total collagen
Total collagen was measured in lung tissue or culture supernatant by biochemical assay (Sircol collagen assay, Biocolor, Belfast, UK) and normalised for tissue weight.
Cytokine analysis
Lung tissue supernatant was analysed by ELISA, IL-4, IL-5, interferon (IFN)-γ (PharMingen, Oxford, UK), IL-33, thymic stromal lymphopoietin (TSLP), CCL20, IL-25, vascular endothelial growth factor (VEGF), activin A and IL-13 kits (R&D systems).8 All data were normalised for lung weight. Paired antibodies for IgE (R&D systems) were used to measure serum IgE levels.
Flow cytometric analysis
Disaggregated lung cells were stained with CD4 andST2 or relevant isotype controls for 20 min at 4°C. Fixed cells were analysed on a FACSCalibur using CellQuest.
SEAP assay
Five microlitres of lung homogenate or standard were added in serum free medium to MFB-F11 cells16 for 24 h prior to measuring bioactivity (Great EscApe SEAP Kit, Clontech, France).
Endothelial progenitor cell colony assay
Following 1 week of HDM exposure lung cell suspensions were separated on discontinuous Percoll gradient. Mononuclear cells were plated in endothelial basal medium (EBM-2/VEGF, 50 ng/ml/17% fetal calf serum (FCS); CambrexBioScience Walkersville Inc, Walkersville, Maryland, USA) on fibronectin-coated dishes. Endothelial progenitor cell (EPC) colonies were scored on day 21 by morphology.17
Fibroblast culture
Normal human lung fibroblasts (NHLFs; Lonza, Basel, Switzerland) were cultured in six well plates in functionally graded materials supplemented with 10% FCS/hygromycin B 15 µg/ml in the presence or absence of 20 ng/ml recombinant human IL-25 (R&D systems). Culture supernatant was analysed for secreted collagen by sircol assay.
Statistical analysis
Data were analysed using Prism 4. Multiple comparisons were performed using the Kruskal–Wallis test. A two-tailed p value was determined by the Mann–Whitney test when comparing between two groups. Data shown are represented graphically as box and whisper plots to depict the median and IQR and minimum and maximum values and are generated from at least two independent experiments (n=4–6 per experiment).
Additional details on the methods used can be found in the online supplementary data file.
Results
Blockade of IL-25 reduces allergen-driven airway remodelling
Elevated levels of IL-25 are observed in mice overexpressing smad2 in the airway epithelium which precedes the development of pronounced airway remodelling in response to HDM exposure.8 We now show that in response to HDM, IL-25 is abundantly produced by airway epithelial cells (figure 1A). Overexpression of smad2 using an adenoviral vector (AdS) or treatment of mice with the control empty vector (AdC) had no effect on pulmonary cellular composition, AHR or parameters of airway remodelling in the absence of allergen exposure.8 Therefore, for clarity, in the current investigation ‘control’ mice are those treated with AdS and PBS.

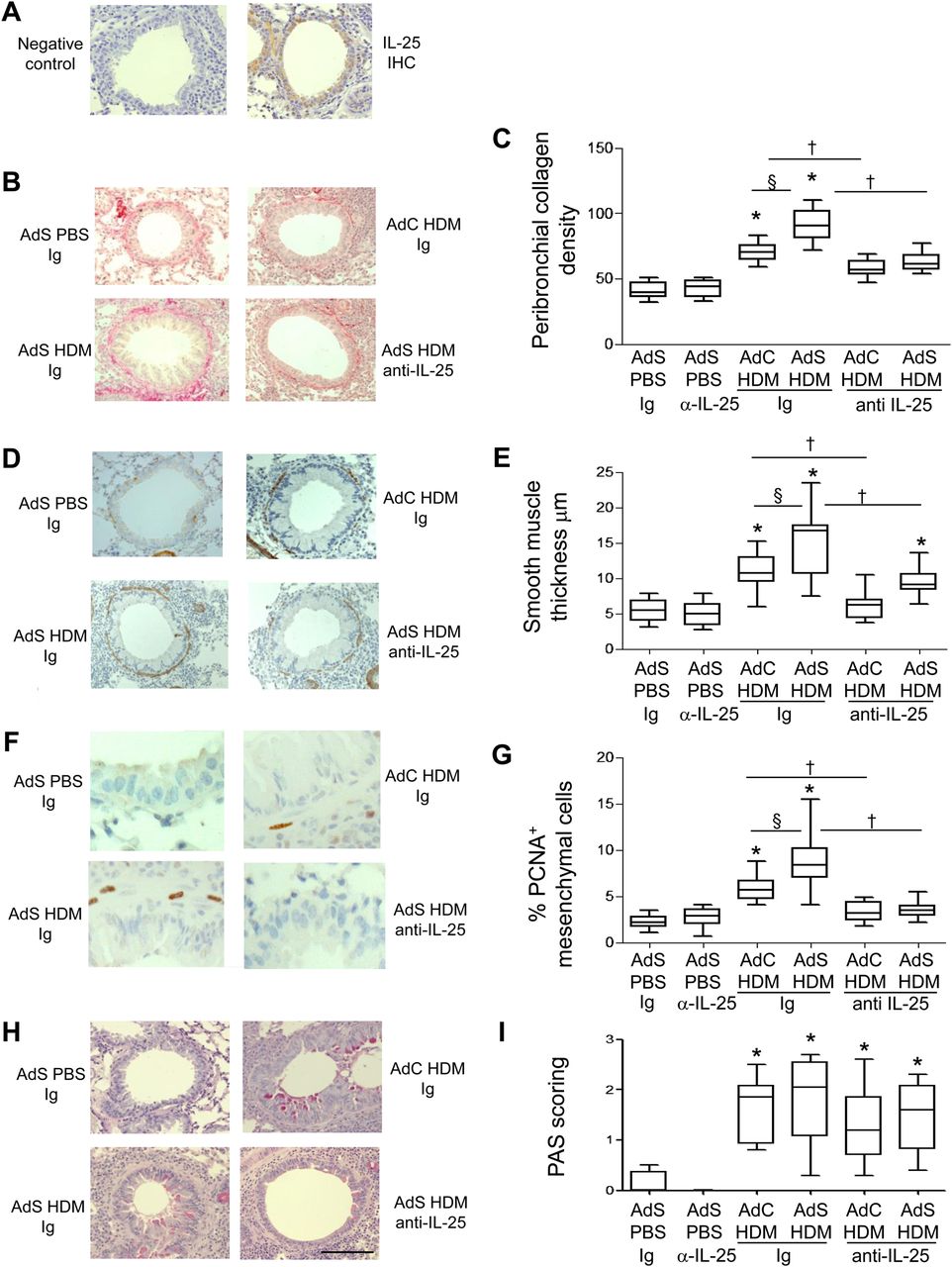
Interleukin (IL)-25 neutralisation abolishes house dust mite (HDM)-induced pulmonary remodelling. (A) IL-25 (brown stained) section. (B) Sirius red stained perivascular and peribronchial collagen. (C) Quantitative analysis of subepithelial peribronchiolar collagen density. (D) α-Smooth muscle actin (brown stained) smooth muscle cells and myofibroblasts. (E) Peribronchial smooth muscle thickness. (F) Proliferating cell nuclear antigen (PCNA)-positive (brown stained) proliferating cells. (G) Percentage of PCNA-positive peribronchial mesenchymal cells. (H) Periodic acid-Schiff (PAS)-positive (pink/purple coloured) mucin-containing cells. (I) Epithelial PAS scoring. Original magnification ×40. Scale bar=50 µm. *p<0.05 compared with phosphate-buffered saline (PBS) control group. §p<0.05 AdS HDM Ig compared with AdC HDM Ig control group. †p<0.05 HDM anti-IL-25 compared with HDM Ig control group. Plots depict the median and IQR and minimum and maximum values. Data are generated from two independent experiments (n=8–12). IHC, immunohistochemistry. This figure is only reproduced in colour in the online version.
Treatment of mice with a neutralising antibody to IL-25 had no effect on basal indices of airway remodelling in the absence of allergen challenge (figure 1). Increased deposition of extracellular matrix is a hallmark feature of airway remodelling and exposure of vector control mice to HDM for 3 weeks resulted in a modest increase in peribronchial collagen (figure 1B,C) which was completely inhibited in mice receiving anti-IL-25 antibody (AdC HDM anti-IL-25) compared with Ig control (AdC HDM Ig). HDM-exposed mice overexpressing epithelial smad2 (AdS HDM) had enhanced collagen deposited around the conducting airways compared with mice receiving allergen alone and even this enhanced de novo matrix deposition was completely abolished by treatment of mice with anti-IL-25 antibody (figure 1B,C).
α-SMA-positive myofibroblasts and smooth muscle cells were increased in HDM-treated mice (figure 1D). Peribronchial smooth muscle thickness was also increased in HDM-challenged mice compared with controls, and this increase was enhanced in mice receiving the AdS vector (figure 1E). However, allergen-induced thickening of the smooth muscle layer was absent in mice administered anti-IL-25 antibody and a significant reduction in ASM mass was observed even in the AdS HDM treated group (figure 1E). Smooth muscle cell proliferation was assessed in lung sections stained with an antibody against PCNA (figure 1F). The percentage of PCNA-positive airway mesenchymal cells was calculated as an index of smooth muscle hyperplasia. This was increased in response to HDM and further elevated in the AdS HDM group (figure 1G). PCNA-positive subepithelial mesenchymal cells were reduced to baseline values in mice pretreated with IL-25 neutralising antibody (figure 1G).
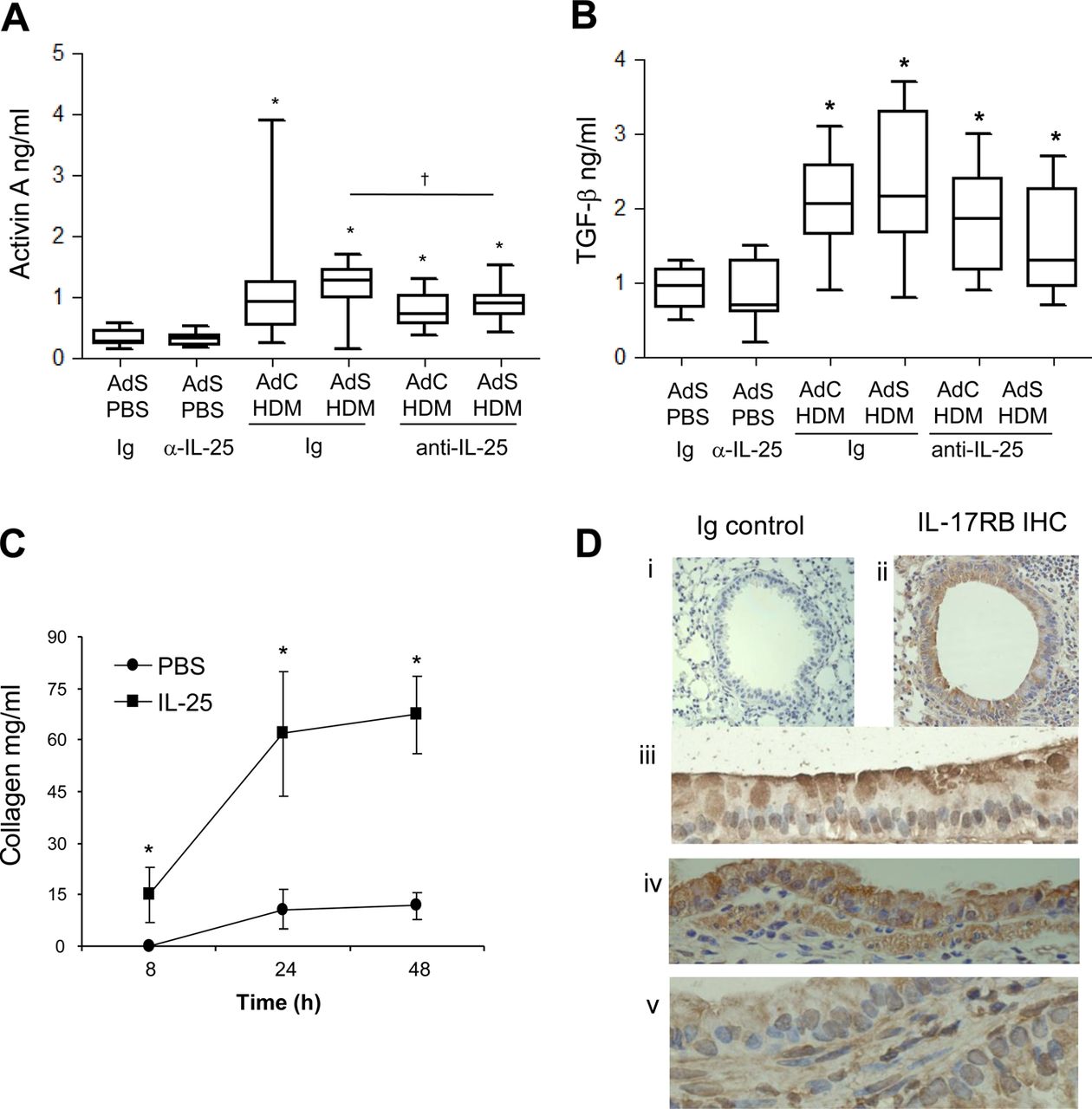
Lung sections were stained with PAS to demonstrate mucus-containing cells (figure 1H). In contrast to the effects on smooth muscle and collagen deposition, neutralising IL-25 did not affect differentiation of the epithelium to a mucus-secreting phenotype (figure 1I). We have previously shown that levels of TGF-β1 and activin A are increased in the lungs of HDM-treated mice and that activin A drives the enhanced remodelling observed in AdS HDM groups.8 We therefore investigated the effect of blocking IL-25 on these two drivers of remodelling. While blocking activin A completely abrogates pulmonary IL-25,8 neutralising IL-25 had only a modest effect on activin A levels (figure 2A) which remain elevated relative to controls. Bioactive TGF-β was elevated in HDM-treated mice but levels were not significantly modulated by prophylactic blockade of IL-25 (figure 2B).

Interleukin (IL)-25 acts directly on mesenchymal cells to induce remodelling. (A) Activin A quantification in lung homogenate. (B) Biologically active pulmonary transforming growth factor (TGF)-β levels. (C) IL-25-induced extracellular collagen secretion by normal human lung fibroblasts (NHLFs) in vitro. (D) IL-17RB (brown stained) lung section: (i) negative Ig control; (ii)–(iv) IL-17RB-positive cells. *p<0.05 compared with phosphate-buffered saline (PBS) control group. §p<0.05 AdS house dust mite (HDM) Ig compared with AdC HDM Ig control group. †p<0.05 HDM anti-IL-25 compared with HDM Ig control group. Plots depict the median and IQR and minimum and maximum values. Data are generated from two independent experiments (n=8–12). IHC, immunohistochemistry. This figure is only reproduced in colour in the online version.
IL-25 acts directly on human mesenchymal cells to induce phenotypic remodelling changes
Blocking IL-25 has profound effects on HDM-induced remodelling, so to determine whether IL-25 acts directly on mesenchymal cells, NHLFs were treated with human recombinant IL-25. This resulted in significantly increased secreted extracellular collagen within 8 h. Collagen synthesis was further increased over 48 h reaching maximal levels by 24 h (figure 2C). These data show for the first time that human fibroblasts respond rapidly to IL-25 by increasing matrix secretion, a hallmark of airway remodelling in human asthma. In the mouse a number of structural cells express the IL-25 receptor (figure 2D), including fibroblasts,18 endothelial cells,19 epithelial cells, particularly at their apical surface (figure 2D iii and iv) and ASM cells (figure 2D v). Thus, IL-25 can potentially act on many structural cells of the lung to directly induce remodelling changes.
IL-25 modulates allergen induced angiogenesis in vivo
Airway wall neovascularisation is a recognised feature of the remodelled asthmatic lung. There was a significant increase in the number of vWF-positive pulmonary blood vessels following exposure of mice to HDM (figure 3A,B). Neutralisation of IL-25 reduced submucosal angiogenesis by 50% (figure 3B). A modest but significant elevation in VEGF was observed in the AdC HDM group but was reduced by treatment of mice with anti-IL-25 antibody (figure 3C). We next investigated recruitment of EPC from the bone marrow to the lung. HDM exposure elicited a significant increase in the number of EPCs in the lung, and this was reduced following IL-25 blockade (figure 3D). Levels of CXCL2, which has been shown to be important for EPC recruitment,14 were elevated in response to HDM challenge and were abrogated in mice administered anti-IL-25 antibody (figure 3E).
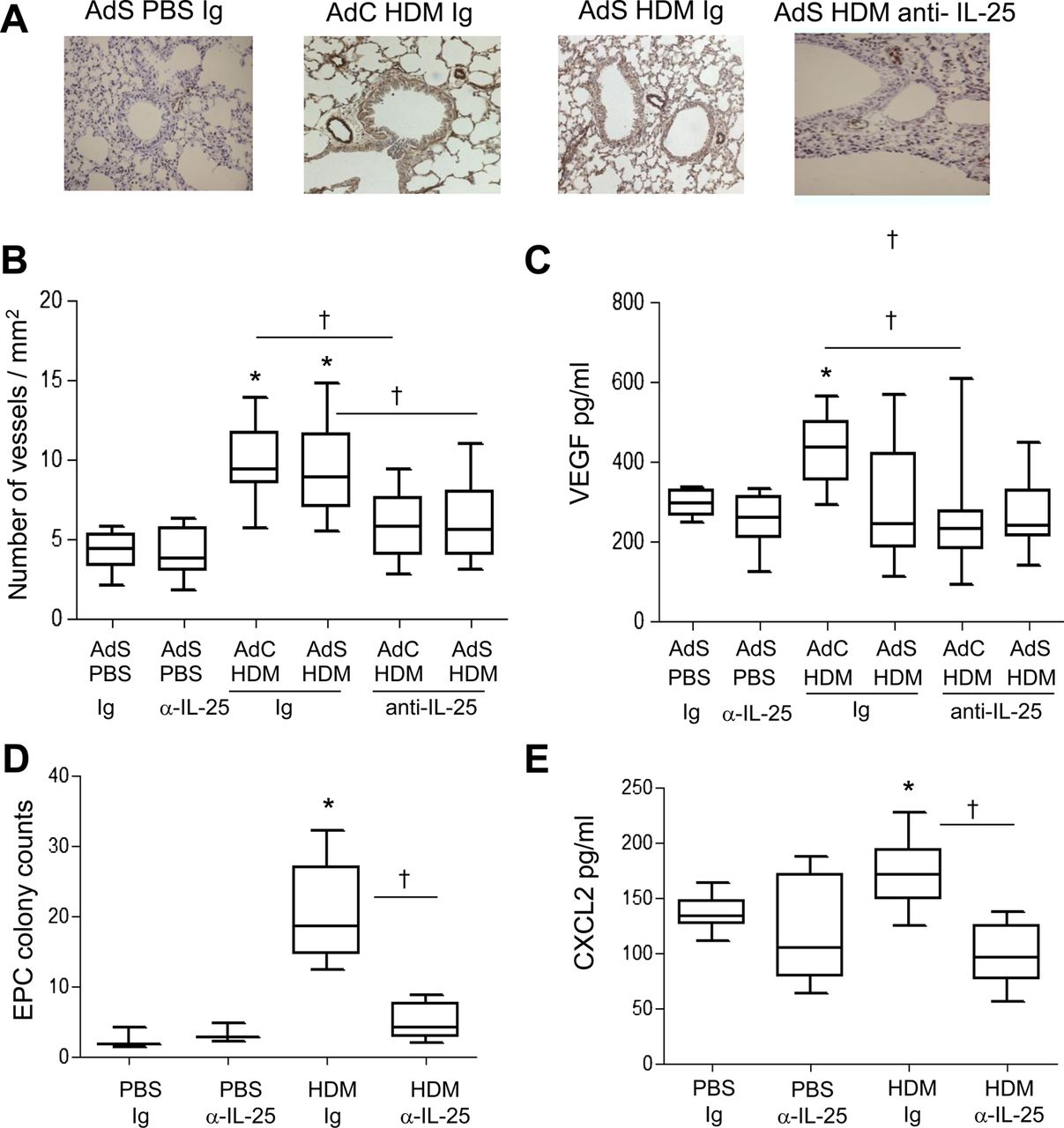
Allergen-induced vascular remodelling. (A) von Willebrand factor (vWF)-positive (brown stained) peribronchial vessels. (B) vWF-positive vessels per square millimetre. (C) Vascular endothelial growth factor (VEGF) levels in the lung homogenate. (D) Pulmonary endothelial progenitor cell (EPC) colony assay. (E) Levels of CXCL2 in lung. *p<0.05 compared with phosphate-buffered saline (PBS) control group. †p<0.05 house dust mite (HDM) anti-interleukin (IL)-25 compared with HDM Ig control group. Plots depict the median and IQR and minimum and maximum values. Data are generated from two independent experiments (n=8–12). This figure is only reproduced in colour in the online version.
IL-25 drives AHR following HDM exposure
IL-25 drives AHR in acute ovalbumin models but we investigated whether IL-25 also promotes the augmented HDM-induced AHR in mice with altered epithelium (figure 4A). This enhanced AHR was reduced by 70% in AdS HDM mice following treatment with anti-IL-25 antibody. AHR was also completely abolished in the AdC HDM treated group following neutralisation of IL-25 (figure 4B). Thus, IL-25 is critical in driving AHR following exposure to an inhaled allergen, even when enhanced by ectopic expression of smad2 in the epithelium.

Airway hyperreactivity is dependent on interleukin (IL)-25. (A) Airway resistance in mice. (B) Airway resistance (RL) at 100 mg/ml methacholine (MCh). *p<0.05 compared with phosphate-buffered saline (PBS) control group. §p<0.05 AdS house dust mite (HDM) Ig compared with AdC HDM Ig control group. †p<0.05 HDM anti-IL-25 compared with HDM Ig control group. Plots depict the median and IQR and minimum and maximum values. Data are generated from two independent experiments (n=8–12).
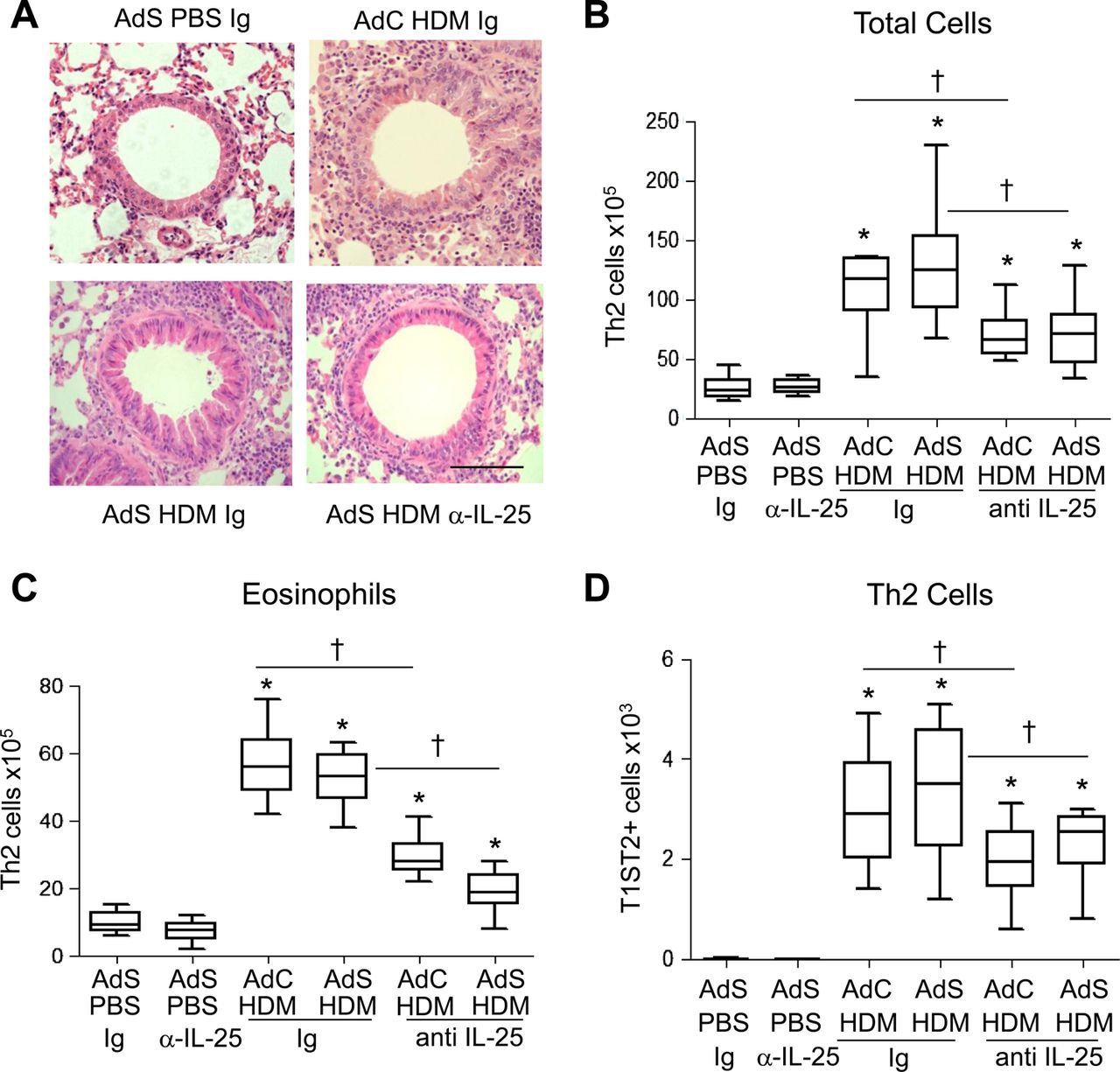
Blocking IL-25 reduces pulmonary inflammation in HDM-exposed mice
Inflammatory cuffs were evident around the conducting airways and blood vessels in H&E-stained lung sections taken from all HDM-treated groups of mice (figure 5A). Mice with altered epithelial smad2 expression had a similar inflammatory response to the AdC groups when challenged with allergen. Treatment of mice with anti-IL-25 antibody resulted in a 50% decrease in the number of recruited cells in the lung (figure 5B) and BAL fluid (data not shown). In particular, eosinophilia, a characteristic feature of asthma, was reduced by anti-IL-25 antibody (figure 5C). Similarly Th2 cells, recruited in response to inhaled HDM, were reduced by blocking IL-25 (figure 5D).

Blocking interleukin (IL)-25 reduces allergen-induced pulmonary inflammation. (A) H&E stained lung sections. Scale bar=50 μm. (B) Total lung inflammatory cells. (C) Pulmonary eosinophil numbers. (D) T helper type 2 (Th2) cells in the lung. *p<0.05 compared with phosphate-buffered saline (PBS) control group. †p<0.05 house dust mite (HDM) anti-IL-25 compared with HDM Ig control group. Plots depict the median and IQR and minimum and maximum values. Data are generated from two independent experiments (n=8–12). This figure is only reproduced in colour in the online version.
IL-25 blockade partially reduces inflammatory mediators in the lung
IL-4, IL-5 and IL-13 were elevated in response to HDM exposure and blocking IL-25 significantly reduced the amount of these cytokines in the lung (figure 6A–C). Levels of IFN-γ, the prototypical Th1 cytokine, are very low in this model of allergic airways disease. However, there was a significant increase in IFN-γ in the AdC PBS anti-IL-25-treated mice (figure 6D). Serum levels of total IgE (figure 6E) and HDM-specific IgE (figure 6F) were elevated following 3 weeks of allergen challenge. Treatment of mice with anti-IL-25 antibody abrogated production of total and antigen-specific IgE.

Allergen-induced inflammatory mediator production is partially decreased by blocking interleukin (IL)-25. (A) Pulmonary IL-4, (B) IL-5, (C) IL-13 and (D) interferon (IFN)-γ levels. (E) Serum total IgE and (F) house dust mite (HDM)-specificIgE determined by optical density (OD). *p<0.05 compared with phosphate-buffered saline (PBS) control group. †p<0.05 HDM anti-IL-25 compared with HDM Ig control group. Plots depict the median and IQR and minimum and maximum values. Data are generated from two independent experiments (n=8–12).
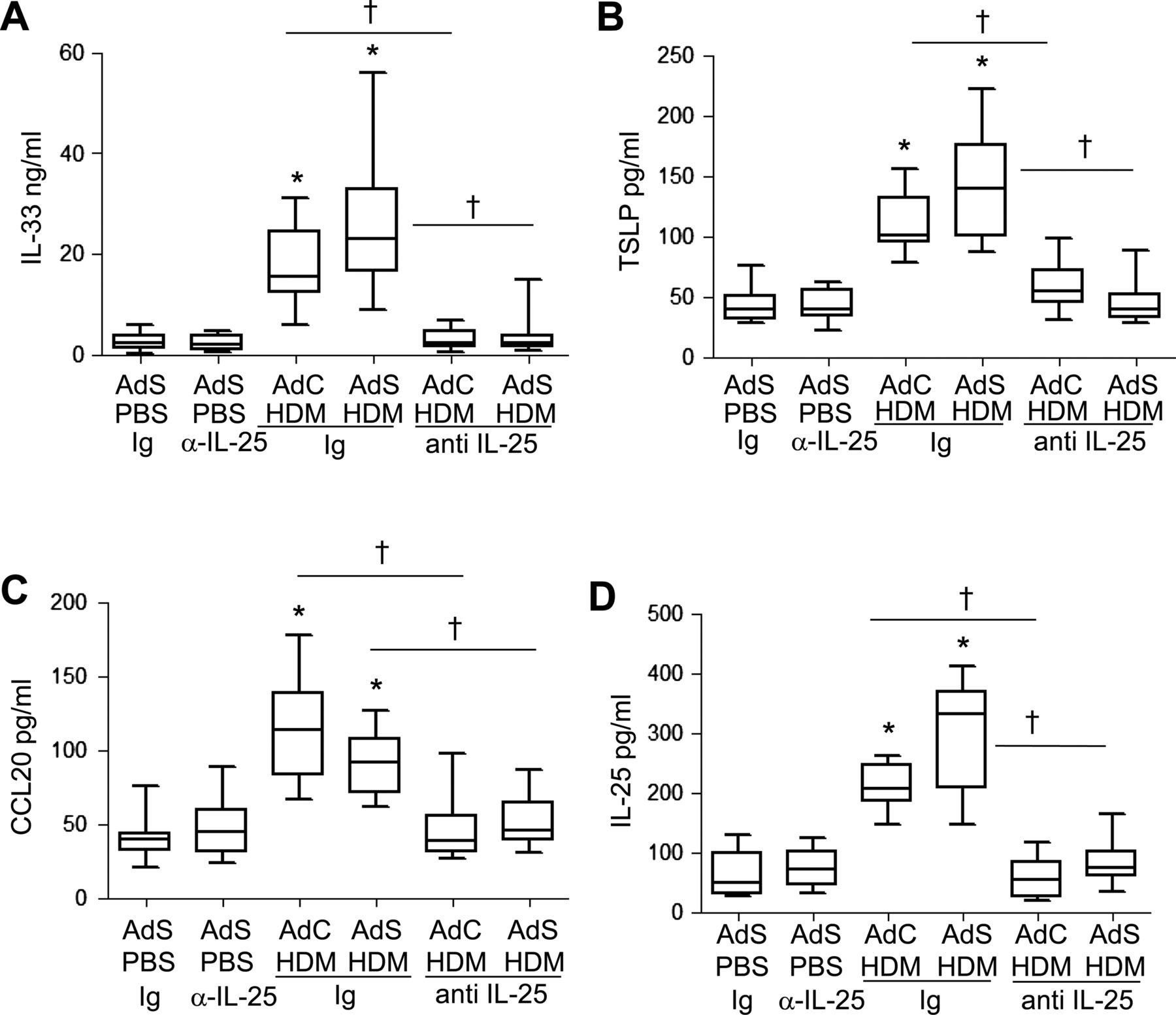
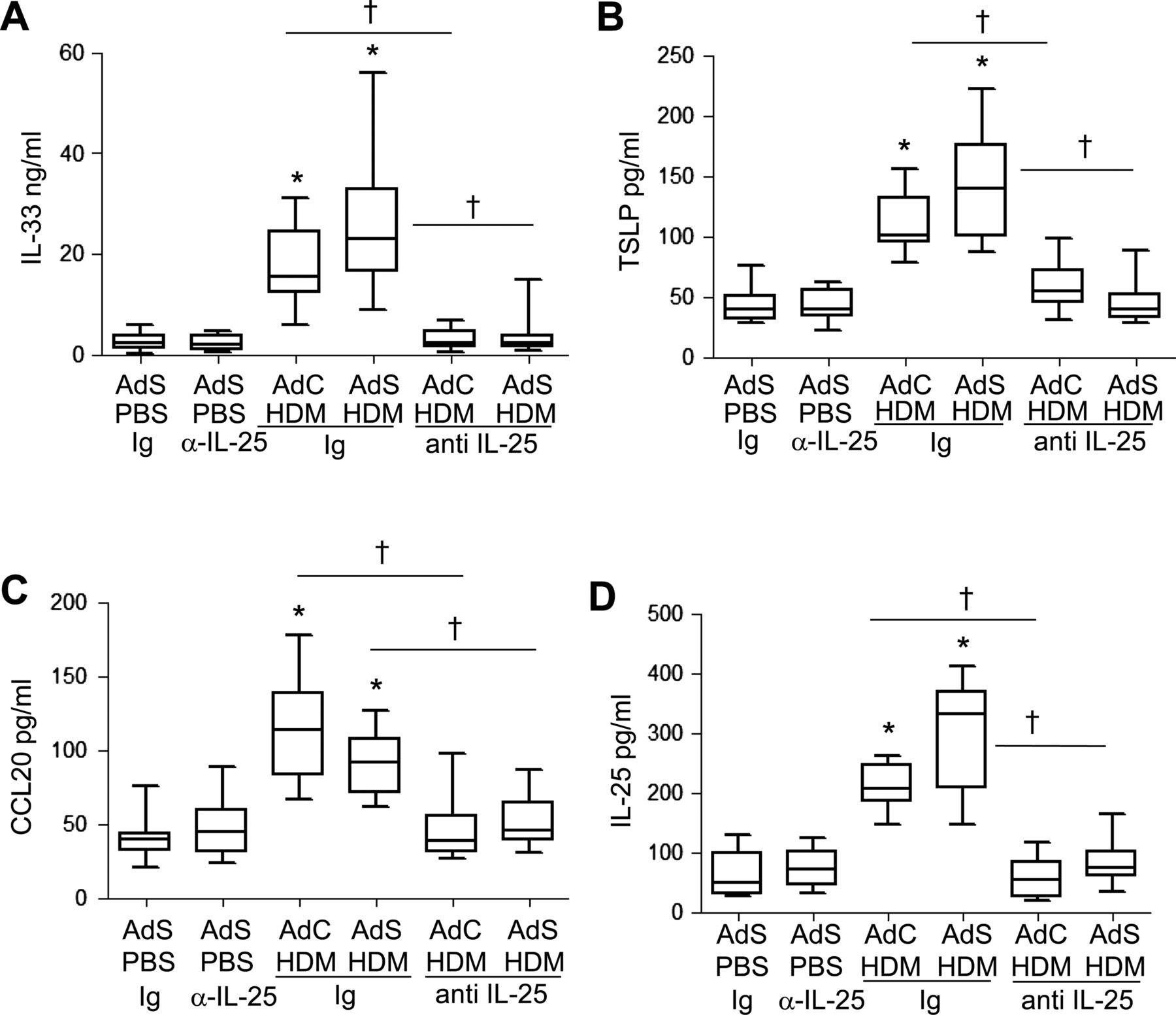
The cytokines IL-25, IL-33, TSLP and CCL20 have been postulated to be important drivers of early allergic Th2 responses in mice, either by the induction of innate helper cells (IL-25 and IL-33) or by differentiation/activation of dendritic cell populations (CCL20 and TSLP). In contrast to the partial effect of blockade of IL-25 on Th2 cytokines associated with the adaptive response, production of the innate epithelial-derived cytokines was totally dependent on the presence of IL-25. IL-33, TSLP, IL-25 and CCL20 were all elevated following 1 week of HDM administration (figure 7A–D), but neutralising IL-25 completely ablated this early innate response to allergen challenge.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Allergen-induced epithelial cytokine release is completely abrogated by blocking interleukin (IL)-25. Levels of (A) IL-33, (B) thymic stromal lymphopoietin (TSLP), (C) CCL20 and (D) IL-25 in the lung. *p<0.05 compared with phosphate-buffered saline (PBS) control group. †p<0.05 house dust mite (HDM) anti-IL-25 compared with HDM Ig control group. Plots depict the median and IQR and minimum and maximum values. Data are generated from two independent experiments (n=8–12).
Collectively, these studies demonstrate that IL-25 is critically involved in airway remodelling in response to HDM. Our data outline a wider role for IL-25 in mediating the chronic and acute effects of allergen exposure.
Discussion
The pathophysiology associated with exposure to allergen is thought to be coordinated by a complex network of mediators including cytokines. We have examined the role of IL-25 in regulating key features of allergic airways disease in response to the clinically relevant allergen HDM. We used mice exposed to allergen alone as well as mice overexpressing smad2 in the airway epithelium, which renders them more susceptible to features of airway remodelling and AHR following HDM exposure.8 Enhanced expression of IL-25, rather than the classical Th2 cytokines IL-13, IL-5 and IL-4, is a prominent feature of this model. These data show that IL-25 plays a critical role in allergen-induced airway remodelling and AHR.
A greater understanding of the molecular mechanisms driving airway remodelling may enhance development of effective treatments. We now show that peribronchial collagen deposition is critically dependent on IL-25. Even in AdS-treated mice, which show enhanced matrix deposition in response to allergen challenge, collagen density is reduced to basal levels when IL-25 is neutralised. To confirm that the observed effects of IL-25 blockade on collagen deposition are a direct effect of IL-25, as opposed to indirect effects induced by IL-25 on the production of Th2 cytokines, NHLFs were treated with IL-25. Fibroblasts, which express the IL-25 receptor subunit IL17RB,18 rapidly upregulated collagen secretion in response to stimulation with IL-25. In addition to fibroblasts we also show in our model that ASM cells express IL-17RB as has previously been reported in patients with asthma.20 It is known that in vitro ASM cells stimulated with IL-25 increase their expression of extracellular matrix components.20 These data show unequivocally that IL-25 can directly induce parameters associated with airway remodelling in human asthma.
Mucus production and goblet cell hyperplasia during an acute ovalbumin model of allergic airways disease are dependent on IL-25.4 In contrast, mice exposed to HDM exhibit epithelial mucus changes independent of IL-25 expression. This is likely driven by eosinophil and T-cell derived IL-13 which was not completely ablated in the lungs of anti-IL-25 HDM-treated mice.
Increased ASM mass is postulated to contribute to altered asthmatic airway function. Mice exposed to HDM exhibit mesenchymal cell hyperplasia and the subsequent increase in ASM mass was completely abrogated following treatment with anti-IL-25 antibody. AHR was concomitantly reduced in allergen-exposed mice treated with IL-25 blocking antibody, even in mice with enhanced AHR following dual exposure with AdS. IL-25 appears to play a critical role in the induction of AHR regardless of the model used to induce allergic airways disease, and this has been shown to be independent of Th2 cytokine production.4
Increased bronchial vascularisation in patients with asthma is thought to contribute to airflow obstruction. Mice exposed to HDM have elevated numbers of pulmonary EPCs and subsequent airway neovascularisation compared with controls. Neutralising IL-25 completely abolished angiogenesis in vivo, confirmed earlier by in vitro observations using human umbilical vein endothelial cells.9 Numbers of EPCs in patients with asthma have been shown to correlate with increased peribronchial blood vessels21 and allergen challenge of patients with atopic asthma results in EPC influx to the airways.22 We have previously shown that EPC recruitment in response to ovalbumin is independent of VEGF A.14 Similarly, levels of VEGF A were not modulated by HDM. However, HDM-induced IL-25-dependent CXCL2 expression correlates with EPC recruitment. Blocking the receptor CXCR2 has been shown to attenuate bronchial angiogenesis in pulmonary ischaemia.23 However, neutralising IL-25 may provide a therapeutic advantage in the treatment of asthma because in addition to preventing neovascularisation other aspects of allergen-induced airway remodelling are also ablated.
TGF-β has been implicated in the development of airway remodelling in ovalbumin-driven asthma models. However, neutralisation of TGF-β during HDM-induced allergic airways disease does not prevent the increase in collagen deposition or α-SMA-positive cells and in fact augments AHR.24 Our present study confirms that HDM-induced airway remodelling is unlikely to be solely driven by TGF-β. In the gut TGF-β acts upstream of IL-2525 but the relationship between TGF-β and IL-25 in the lung has yet to be fully elucidated. Our previous work has shown that the TGF-β family member activin A is also involved in the development of HDM-driven airway remodelling. Neutralising activin A abrogated the HDM-induced increase in pulmonary IL-25 levels.8 Blocking IL-25 had a modest effect on activin A, however levels remained significantly elevated compared with control mice. We have previously shown that it is necessary to block TGF-β and activin A to prevent pulmonary allergic pathology.26 Our current data suggest that activin A acts upstream of IL-25 and that both cooperate to promote airway remodelling.
Overexpression of IL-25 results in elevated IL-4, IL-5 and IL-13 gene expression and serum IgE, IgG1 and IgA levels in addition to eosinophilia, increased mucus production and epithelial cell hyperplasia/hypertrophy.27 Administration of anti-IL-25 antibody to HDM-exposed mice significantly decreased pulmonary inflammation and Th2 cytokine production, although levels remained elevated compared with PBS control mice. In contrast, IL-25 neutralisation completely abrogated the allergen-induced early increases in the innate cytokines, IL-33 and TSLP. TSLP, IL-25 and IL-33 are an epithelial-derived triad of cytokines which collectively drive Th2 polarisation through complementary and sometimes synergistic mechanisms. Emerging evidence suggests that IL-33 and TSLP may also be associated with tissue remodelling. Expression of IL-33 is strongly associated with fibrosis in chronic liver injury and skin and IL-33 levels have been shown to correlate with pulmonary fibrosis and decreased forced vital capacity.28 ,29 Intradermal administered TSLP promotes subcuticular fibrosis in mice and TSLP can enhance fibrocyte differentiation and increase collagen production.30 ,31 Of interest, IL-25 has been shown to increase TSLP expression in MLE12 epithelial cells,1 and we have shown that murine airway epithelial cells express IL-17RB. Our data suggest that IL-25 drives production of IL-33 and TSLP in vivo and might also cooperate with these cytokines to orchestrate airway remodelling, confirming a central role of the airway epithelium in driving the pathological changes observed in the remodelled asthmatic lung.
In conclusion, neutralising IL-25 has a limited effect on recruitment of leukocytes to the lung when a complex allergen is used. In contrast, blocking IL-25 completely inhibits the release of the epithelial-derived innate cytokines and abrogates subepithelial fibrosis and mesenchymal cell proliferation and AHR. Focusing specifically on mediators which play a crucial role in driving remodelling and AHR presents a novel therapeutic approach to the treatment of asthma, particularly as this aspect of disease pathology is resistant to most current therapies.
Acknowledgments
The authors thank Lorraine Lawrence for histological sectioning and staining.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors LGG conducted the majority of the experiments, analysed the data and wrote the manuscript; CPJ, SAW, DS and KG performed some of the experimental work; ANJM produced the anti-IL-25 antibody and reviewed the manuscript; CML designed the study, supervised the project and edited the article.
-
Funding This work was supported by the Welcome Trust (grants 057704 and 085851/Z/08/Z).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement There are no additional unpublished data.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/