Article Text

Abstract
Background Therapeutic strategies to modulate the host response to bacterial pneumonia are needed to improve outcomes during community-acquired pneumonia. This study used mice with impaired Fas signalling to examine susceptibility to pneumococcal pneumonia and decoy receptor 3 analogue (DcR3-a) to correct factors associated with increased susceptibility.
Methods Wild-type mice and those with varying degrees of impairment of Fas (lpr) or Fas ligand signalling (gld) were challenged with Streptococcus pneumoniae and microbiological and immunological outcomes measured in the presence or absence of DcR3-a.
Results During established pneumonia, neutrophils became the predominant cell in the airway and gld mice were less able to clear bacteria from the lungs, demonstrating localised impairment of pulmonary neutrophil function in comparison to lpr or wild-type mice. T-cells from gld mice had enhanced activation and reduced apoptosis in comparison to wild-type and lpr mice during established pneumonia. Treatment with DcR3-a reduced T-cell activation and corrected the defect in pulmonary bacterial clearance in gld mice.
Conclusions The results suggest that imbalance in tumour necrosis factor superfamily signalling and excessive T-cell activation can impair bacterial clearance in the lung but that DcR3-a treatment can reduce T-cell activation, restore optimal pulmonary neutrophil function and enhance bacterial clearance during S pneumoniae infection.
- Streptococcus pneumoniae
- pneumococcal pneumonia
- aFas/Fas ligand
- T-cell activation
- murine models of pneumococcal infection
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Streptococcus pneumoniae
- pneumococcal pneumonia
- aFas/Fas ligand
- T-cell activation
- murine models of pneumococcal infection
Key messages
What is the key question?
-
Does tumour necrosis factor (TNF) superfamily member signalling modify outcomes of pneumococcal pneumonia?
What is the bottom line?
-
The balance of Fas ligand and decoy receptor 3 inhibitable ligands influences T-cell activation and local neutrophil function in the lung during pneumococcal pneumonia.
Why read on?
-
Immunomodulation of TNF superfamily signalling has potential as immunotherapy during community-acquired pneumonia.
Introduction
Streptococcus pneumoniae (the pneumococcus) is the commonest cause of community-acquired pneumonia (CAP) and causes approximately 2 million deaths annually.1 Despite advances in antimicrobial therapy and management of critically ill patients, therapeutic modulation of the pulmonary inflammatory response has been relatively neglected in CAP, even though dysregulation of the inflammatory response is a key feature of pathogenesis. To adopt this strategy we need a better understanding of factors which regulate susceptibility to CAP, and how these can be manipulated to ensure an optimal inflammatory response that achieves bacterial clearance without inflammatory lung injury.
The tumour necrosis factor superfamily (TNFSF) regulates inflammatory responses. In this study we have addressed whether TNFSF member Fas ligand (FasL, TNFSF6) influences susceptibility to pneumococcal pneumonia. FasL is a transmembrane protein that signals through Fas (CD95, TNFRSF6).2 Multimerised Fas activates apoptosis in a range of cells, including epithelial cells and phagocytes, regulating immune homeostasis.3 Alternatively Fas signal transduction contributes to cell activation and pro-inflammatory effects, for example in the lung FasL contributes to the pathogenesis of acute lung injury.4 FasL expression increases during pneumococcal infection.5 ,6 During pneumonia with Gram-positive bacteria there is less neutrophil recruitment in lpr mice, which have low levels of functional Fas, or in mice treated with a decoy receptor 3 analogue (DcR3-a) that neutralises FasL.6 ,7 Despite this, bacterial clearance is enhanced unless a more severe infection is established, suggesting under certain circumstances FasL has an adverse effect on disease outcome.6 FasL therefore has key roles in immune homeostasis,8 and host defence.9 It also has the potential to exacerbate inflammatory lung injury, and its role in bacterial clearance varies depending on the infection and murine model studied.6 ,7
We addressed the role of FasL in the pathogenesis of CAP in murine models of pneumococcal pneumonia (see online supplement). We tested the role of complete FasL deficiency using C57BL/6 gld mice, which express FasL lacking functional activity.10 The results were compared with wild-type C57BL/6 mice and C57BL/6 mice carrying the lpr mutation, which results in reduced expression of Fas,11 but less than complete inhibition of Fas signalling, since the mutation is leaky.12 ,13 We also compared results in these mice with the effect of FasL neutralisation with DcR3-a. DcR3-a is an analogue of DcR3 (TNFRSF6B) which neutralises FasL but also inhibits two other TNFSF members: TNFSF14 (lymphotoxin-related inducible ligand which competes for glycoprotein D binding to herpes virus entry mediator on T cells; LIGHT) and TNFSF15 (TNF-like protein 1A; TL1A).14–16 This allowed us to compare the effects of selective versus non-selective FasL neutralisation that also included other TNFSF members to determine their potential role in the pathogenesis of CAP and to examine the therapeutic potential of manipulation of TNFSF signalling during CAP.
Our murine models suggest inhibition of FasL signalling enhances T-cell activation and impedes bacterial clearance but that these alterations can be corrected by administration of a TNFSF decoy receptor 3 analogue (DcR3-a).
Material and methods
Animals
Adult female mice, homozygous for the FasLgld mutation (B6Smn.C3-Faslgld/J) and Faslpr(B6.MRL-Faslpr/J), on a C57BL/6 background (Jackson Laboratories, Bar Harbour, Maine, USA) were used.10 ,11 C57BL/6 mice were used as wild-type controls. All animal experiments were conducted in accordance with the Home Office Animals (Scientific Procedures) Act 1986 and approved by the University of Sheffield Ethical Review Committee.
Pneumococcal infection model
Pulmonary infection of mice was by direct tracheal instillation as described previously.17 Neutrophil depletion was with 100 μg/mouse Gr-1 antibody or isotype control. Decoy receptor treatment was with 400 μg/mouse DcR3-a or bovine serum albumin (BSA). For detailed methods see online supplement. Viable bacteria in lung and blood were measured and cell differential was assessed by morphology on cytospins.17 Apoptosis detection was by nuclear morphology or Annexin V—TopPro 3 staining and flow cytometry, with neutrophils being identified by Ly6G staining and forward/side scatter characteristics as described.17
In vitro neutrophil function
Peripheral blood neutrophils were isolated by negative magnetic selection with cell purity routinely greater than 80%.18 Rates of apoptosis were assessed by morphology on cytospins after 9 h of culture. For apoptosis after bacterial infection neutrophils were infected with serotype 2 S pneumoniae (multiplicity of infection (MOI) =10) and apoptosis assessed at 9 h post infection by nuclear morphology. Respiratory burst was assessed by flow cytometry analysis of dichlorodihydrofluorescein fluorescence following 30 min of stimulation by heat-killed serotype 2 S pneumoniae (MOI=10). Phagocytosis was assessed by calculating the phagocytic index on cytospins of neutrophils incubated with opsonized zymosan (20 μg/ml) for 30 min; internalised particles were counted as those within a phagocytic vacuole.
Isolation and staining of splenocytes
Mice received 1×107 colony-forming units (CFUs) of serotype 4 S pneumoniae and after 24 h were killed by overdose of anaesthetic and exsanguination. Spleens were collected, splenocytes isolated and activation of CD3+ T cells measured by flow cytometry (LSR II, BD Biosciences, Oxford, UK) as percentage of CD25+ cells. Apoptosis was measured as the percentage of cells with hypodiploid DNA after propidium iodide staining.19 Analysis was with FlowJo software V.9.3.2 (Tree Star, Inc, Ashland, Oregon, USA).
Statistics
Statistical analysis was by analysis of variance (parametric data), t test (parametric data), Kruskal–Wallis (non-parametric data), Mann–Whitney (non-parametric data) or log-rank test for survival, using Prism 5.0 software (GraphPad Inc, La Jolla, California, USA). Differences were considered significant if p<0.05.
Results
Reduced bacterial clearance in gld mice with established pneumonia
We examined the outcome of infection using three models of infection with high inocula of S pneumoniae. There was an increase in CFUs in lung and blood, 24 h after infection, in the gld mice compared with C57BL/6 and lpr mice (which had similar CFUs), after both serotype 1 (figure 1A,B) and serotype 2 infection (figure 1D,E). Serotype 4 infections resulted in higher bacterial CFUs in lung or blood of wild-type and lpr mice but the increase in CFUs for gld mice was not significant (figure 1G,H).

Reduced bacterial clearance in gld mice with established pneumonia. (A) Colony-forming units (CFUs) of bacteria in lung homogenates from wild-type control mice (WT) and mice deficient in Fas ligand (gld) or Fas (lpr) 24 h after intratracheal instillation of 107 CFUs of serotype 1 pneumococci; (B) CFUs of bacteria in blood in the same experiments as (A); (C) percentage of neutrophils (PMN) in bronchial alveolar lavage (BAL) from the same experiments as (A); (D) CFUs of bacteria in lung homogenates from mice 24 h after intratracheal instillation of 107 CFUs of serotype 2 pneumococci; (E) CFUs of bacteria in blood in the same experiments as (D); (F) percentage of neutrophils in BAL from the same experiments as (D); (G) CFUs of bacteria in lung homogenates from mice 24 h after intratracheal instillation of 107 CFUs of serotype 4 pneumococci; (H) CFUs of bacteria in blood in the same experiments as (G); (I) percentage of neutrophils in BAL from the same experiments as (G). (A, B, D, E, G, H) Line at median, Kruskal–Wallis test with Dunn's multiple comparison test; (C, F, I): mean+SEM, analysis of variance with Bonferroni's multiple comparison test, *p<0.05, **p<0.01, ***p<0.001, n=12–15.
FasL contributes to CXC chemokine expression in the lung and to neutrophil recruitment into the airspaces in response to a variety of stimuli including pneumococci.6 ,20 We documented impaired neutrophil recruitment into the lung in the absence of Fas signalling in both gld and lpr mice 24 h after challenge with serotype 1 S pneumoniae (figure 1C). After infection with the more virulent serotypes 2 or 4 S pneumoniae, neutrophil numbers were similar between strains at 24 h (figure 1F,I). Fas can trigger neutrophil apoptosis.3 Since absolute levels of neutrophil apoptosis in vivo in the lung are influenced by the rate of apoptosis induction and the rate of clearance of apoptotic cells, while the percentage of apoptotic cells is confounded by rates of neutrophil recruitment, we measured rates of constitutive apoptosis in vitro. We confirmed reduced rates of constitutive apoptosis in peripheral blood neutrophils from lpr and gld mice, but found no reduction in neutrophil apoptosis in the presence of pneumococci in vitro and confirmed this finding in vivo (figure 2A–C).
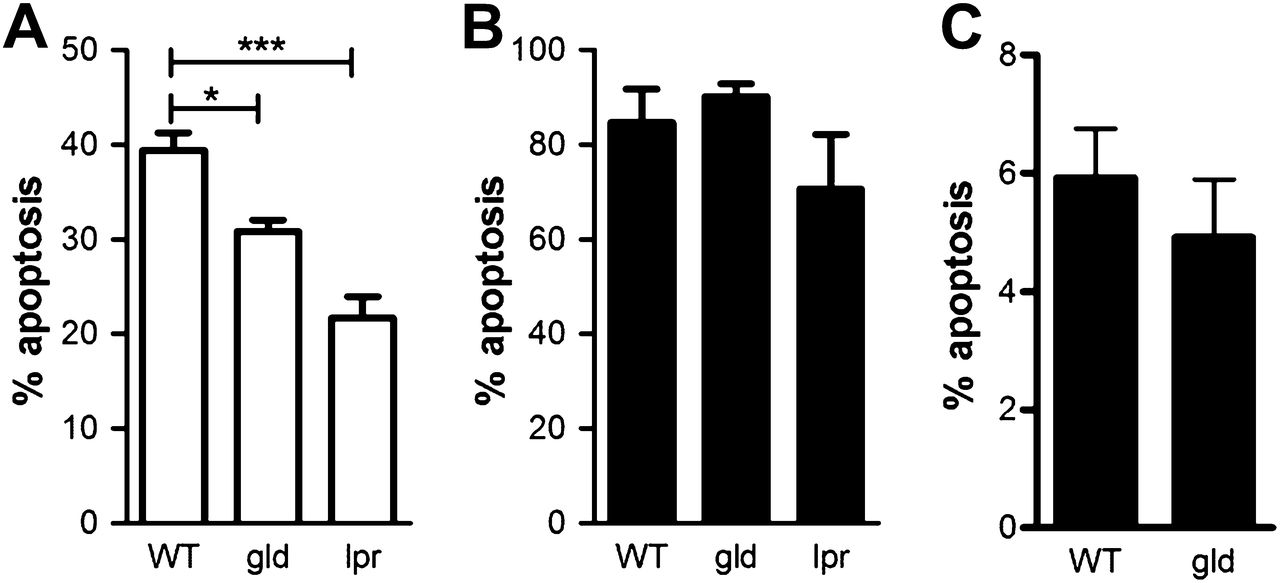
Mice lacking Fas ligand have reduced constitutive neutrophil apoptosis but no defect in apoptosis in the presence of bacteria. Peripheral blood neutrophils from wild-type control mice (WT) and mice deficient in Fas ligand (gld) or Fas (lpr) were isolated by negative magnetic selection. (A) Rates of apoptosis were assessed by morphology on cytospins prepared after 9 h of culture, n=4. (B) Neutrophils were infected with pneumococci (MOI=10) and apoptosis assessed as in A, n=3. (C) Apoptosis of neutrophils in bronchial alveolar lavage from WT control mice or mice deficient in Fas ligand (gld) 24 h after intratracheal instillation of 107 colony-forming units (CFUs) of serotype 2 pneumococci, n=14 WT and 10 gld. Mean+SEM, analysis of variance with Bonferroni's multiple comparison test, *p<0.05, ***p<0.001.
Dysfunctional pulmonary neutrophil responses in gld mice
We next addressed the functional capacity of phagocytes. As gld mice had no reduction in alveolar macrophage-dependent bacterial clearance (online supplementary figure 3) we addressed the functional capacity of neutrophils. Since the number of bacteria inside neutrophils reflects rates of ingestion and killing we measured rates of zymosan uptake as a marker of phagocytic capacity. Peripheral blood neutrophils from gld and lpr mice had no intrinsic defect in phagocytosis or in generation of reactive oxygen species when challenged with heat-killed S pneumoniae ex vivo (figure 3A,B). To address the contribution of neutrophils to bacterial clearance in vivo we reduced neutrophil numbers with anti-Gr1 antibody (figure 4A, online supplementary figure 2) and examined clearance of serotype 2 S pneumoniae at 14 h after infection, to ensure the phagocytes were recently recruited.21 Although anti-Gr-1 also depletes monocytes these made up only about 1% of bronchial alveolar lavage cells at this time point. The anti-Gr-1 treatment significantly increased CFUs in the lung and blood of C57BL/6 and lpr mice but not gld mice (figure 4B,C). Since the defect in bacterial CFUs was more marked for gld mice in blood than in lung we also investigated whether these mice were less able to clear bacteria from blood. A tail vein injection of 103 CFUs showed equivalent bacterial clearance in C57BL6 and gld mice (data not shown).

Mice lacking Fas ligand have preserved neutrophil function ex vitro. Peripheral blood neutrophils from wild-type control mice (WT) and mice deficient in Fas ligand (gld) or Fas (lpr) were isolated by negative magnetic selection. (A) Phagocytosis was assessed by calculating phagocytic index on cytospins of peripheral blood neutrophils incubated with opsonized zymosan (20 μg/ml) for 30 min, n=5. (B) Respiratory burst was assessed by flow cytometry analysis of dichlorodihydrofluorescein (DCF) fluorescence following 30 min of stimulation by heat-killed serotype 2 pneumococci (MOI=10). Geometric mean fluorescence (GMF), n=3. Mean+SEM, analysis of variance with Bonferroni's multiple comparison test, p = not significant.
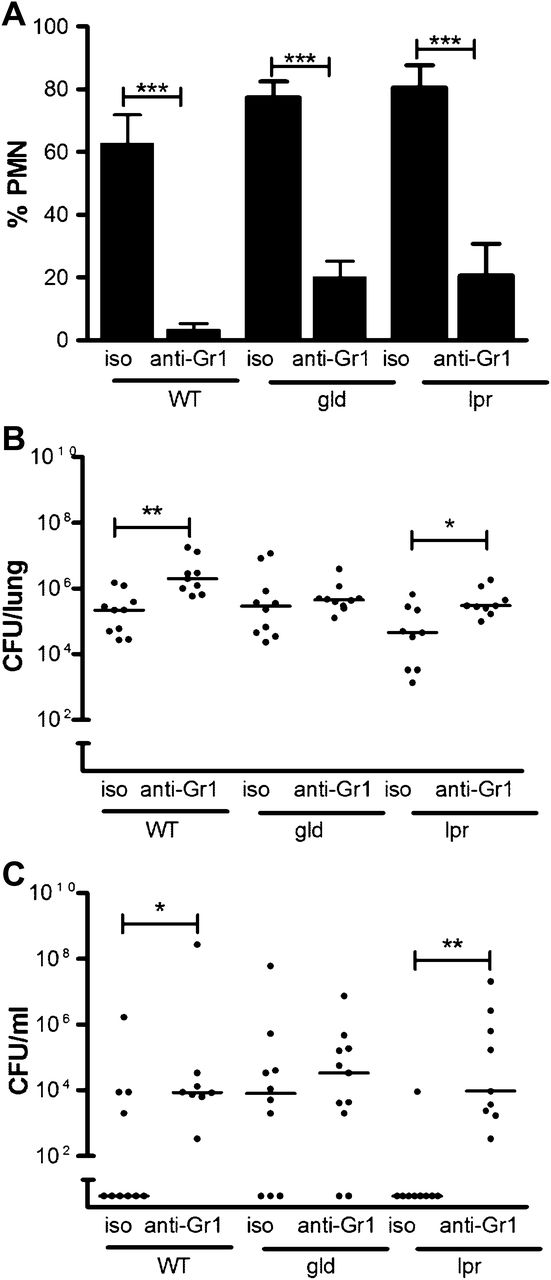
Absence of Fas ligand impairs neutrophil function in vivo. Wild-type control mice (WT) and mice deficient in Fas ligand (gld) or Fas (lpr) were treated with anti-Gr1 antibody to deplete neutrophils (PMN) (anti-Gr1) or isotype control (iso) 24 h prior to intratracheal instillation of 107 colony-forming units (CFUs) of serotype 2 pneumococci. Fourteen hours post infection (A) the percentage of neutrophils in bronchial alveolar lavage was assessed, mean+SEM, analysis of variance with Bonferroni's multiple comparison test; (B) CFUs of bacteria in lung homogenates; (C) CFUs of bacteria in blood, line at median, Kruskal–Wallis test with Dunn's multiple comparison test, *p<0.05, **p<0.01, ***p<0.001, n=9–10.
Results at 48 h after high-dose serotype 2 infection also revealed increased CFUs in gld mice (figure 5A,B) though to a lesser extent than at 24 h (figure 1D,E), with reduced neutrophil counts (figure 5C). In keeping with these findings gld mice had increased mortality in comparison to wild-type mice in the period from 72 to 120 h (figure 5D). At 72 h there was 40% survival in the gld mice versus 70% in the wild-type mice and the median survival in gld mice was 72 h versus undefined in the wild-type mice, although overall survival did not differ (p=0.5033).

The difference in bacterial outcome between wild-type (WT) mice and mice deficient in Fas ligand (gld) is reduced at later time points. (A) Colony-forming units (CFUs) of bacteria in lung homogenates from WT control mice (n=14) and gld mice (n=12) 48 h after intratracheal instillation of 1×107 CFUs of serotype 2 pneumococci. (B) CFUs of bacteria in blood in the same experiments as (A), line at median, Mann–Whitney. (C) The percentage of neutrophils (PMN) in bronchial alveolar lavage in the same experiments as (A), mean+SEM, t test, **p<0.01. (D) WT (n=11) and gld mice (n=12) were instilled with 107 CFUs of type 2 pneumococci and followed for 10 days. Median survival: WT, undefined; gld, 72 h; log-rank test p=0.5033.
Alterations in the levels of T-cell activation and apoptosis in gld mice
Since accumulation of activated T cells is an early feature of pneumococcal pneumonia and T-cell responses are required for optimal clearance of bacteria,22 ,23 we next examined levels of T-cell activation in each mouse strain by studying levels of CD25+ CD3+ T cells in cells isolated 24 h after infection. We used high-dose type 4 S pneumoniae to ensure all mice had equivalent levels of bacteraemia. As shown there were greater levels of CD3+ T-cell activation in gld mice (figure 6A) and these mice had lower levels of apoptotic T cells (figure 6B).

Alterations in levels of T-cell activation and apoptosis in mice deficient in Fas ligand (gld). Splenocytes were isolated from spleens of wild-type control mice (WT) and gld mice or mice deficient in Fas (lpr) 24 h after intratracheal instillation of 107 colony-forming units of serotype 4 pneumococci and CD3+ T cells identified by flow cytometry. (A) Activation was measured by the levels of CD25 cells (n=3). (B) Apoptosis was measured by analysing the percentage of cells with hypodiploid DNA after propidium iodide staining (% Sub G0/1, n=5), mean+SEM, analysis of variance with Bonferroni's multiple comparison test, *p<0.05.
DcR3-a corrects the impairment in bacterial clearance associated with FasL deficiency
The decoy receptor DcR3 not only neutralises FasL but also additional TNFSF members LIGHT and TL1A and can modify T-cell activation.24 ,25 We reasoned if the impaired clearance of bacteria was solely due to FasL deficiency then DcR3 would replicate the gld phenotype in C57BL/6 and lpr mice but have no effect upon gld mice. To test this we utilised a modified analogue of DcR3, DcR3-a (FLINT), which is more resistant to proteolytic cleavage in vivo than the parental structure.6 ,26 DcR3-a inhibits FasL, LIGHT and TL1A in vivo and has modified bacterial clearance in other models of pneumococcal pneumonia.6 ,26 Surprisingly we did not find any difference in bacterial CFUs in the lungs or blood of C57BL/6 and lpr mice in the presence or absence of DcR3-a (figure 7A,B) following infection with a moderate dose of serotype 4 pneumococci, which resulted in only low levels of neutrophil recruitment (figure 7C). In contrast, the defect in bacterial clearance was corrected in the gld mice. DcR3-a also reversed the increased levels of T-cell activation observed in gld mice (figure 8).

Decoy receptor 3 analogue (DcR3a) corrects the impairment in bacterial clearance associated with Fas ligand deficiency. Wild-type control mice (WT) and mice deficient in Fas ligand (gld) or Fas (lpr) were treated with DcR3a or BSA control (BSA) immediately prior to intratracheal instillation of 5×105 colony-forming units (CFUs) of serotype 4 pneumococci and 12 h post infection. Twenty-four hours post infection, (A) CFUs of bacteria in lung homogenates. (B) CFUs of bacteria in blood in the same experiments as (A), line at median, Kruskal–Wallis test with Dunn's multiple comparison test. (C) The percentage of neutrophils (PMN) in bronchial alveolar lavage in the same experiments as (A) and (B) were estimated, mean+SEM, analysis of variance with Bonferroni's multiple comparison test, *p<0.05, n=8–16.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
T-cell activation is reduced in mice deficient in Fas ligand (gld) after decoy receptor 3 analogue (DcR3a) treatment. Wild-type control mice (WT) and gld mice were treated with DcR3a or BSA control (BSA) immediately prior to intratracheal instillation of 1×107 colony-forming units (CFUs) of serotype 4 pneumococci. Splenocytes were isolated from spleens 24 h after infection, and the percentage of CD25+ CD3+ T cells recorded by flow cytometry, mean+SEM, analysis of variance with Bonferroni's multiple comparison test, *p<0.05, n=3–6.
Discussion
We demonstrate that gld mice are impaired in their ability to clear bacteria from the lung in a model of pneumococcal pneumonia. We show enhanced T-cell activation is associated with localised pulmonary defects in neutrophil function in gld mice. DcR3-a, which neutralises FasL, had no effect on bacterial clearance in wild-type or lpr mice but corrected the defects in bacterial clearance in gld mice and reduced levels of T-cell activation, suggesting unopposed T-cell activation by other TNF superfamily members, inhibitable by DcR3-a, influenced bacterial clearance in gld mice. These results suggest that altered balance of TNFSF signalling, involving FasL and other DcR3 ligands, adversely influences the outcome of pneumonia during a defined period when initial neutrophil and T-cell recruitment are required to control infection and at a defined threshold of infection when resident host responses fail.22 ,23
The effects of complete and selective inhibition of Fas signalling on pneumococcal pneumonia, to our knowledge, have not previously been addressed. We did not predict a difference between gld and lpr mice. Both mutations are loss of function mutations that impair Fas signalling leading to autoimmune manifestations13 and we confirmed the development of lymphadenopathy and splenomegaly as gld and lpr mice aged. The extent of inhibition of Fas signalling in lpr mice is less complete than that observed in gld mice.12 ,13 The lpr mutation results from the insertion of an early transposable element into intron 2 and is leaky with residual low-level expression of Fas.12 In contrast, the gld mutation introduces a point mutation into the C-terminus of FasL and blocks binding of FasL to Fas.10 These result in subtle differences in the inflammatory response between strains.20 Of immediate relevance gld mice have a more profound reduction in peripheral blood CD4+, CD4+ CD25+ FoxP3+, and most markedly CD8+ T cells.27 We found reduced neutrophil-dependent bacterial clearance and enhanced T-cell activation occurred only in gld mice, suggesting residual Fas signalling activity in lpr mice may have been sufficient to ensure optimal host responses and to prevent the more marked perturbations in T-cell subsets previously described in gld mice.27
We show an early requirement for FasL at the stage of pneumonia when neutrophils start to be recruited to the lung. Under these circumstances absolute and selective deficiency of FasL was associated with enhanced activation of T cells and reduced pulmonary neutrophil function. Residual Fas signalling, as evidenced by lpr mice, is sufficient for host defence but complete deficiency exposes the host to harmful effects of other TNFSF members. The concept that the unopposed actions of other TNFSF members may mediate specific phenotypes during impairment of Fas signalling is not without precedent since neutralisation of TNF-α attenuates the generalised lymphadenopathy of gld mice.28
Fas signalling influenced neutrophil recruitment and rates of constitutive apoptosis, as previously documented.6 It did not alter rates of neutrophil apoptosis after bacterial infection. Overall this resulted in variable differences in neutrophil numbers in the lung. Differences in neutrophil numbers alone do not explain the selective impairment in host defence of the gld mice and there was no intrinsic deficit in neutrophil function in either lpr or gld mice. The functional neutrophil impairment in bacterial clearance was confined to the lung since clearance from the blood was normal following intravenous challenge. We conclude that there was a localised defect in the phagocytosis or killing of bacteria by pulmonary neutrophils in gld mice. Fas signalling can modify toll-like receptor responses27 but the absence of macrophage or intrinsic neutrophil defects argues against this being an important factor. The unique defects we identified in gld mice were characterised by selective alteration in pulmonary neutrophil function and were associated with increased T-cell activation.
The role of T cells in the innate response to pneumococcal pneumonia has been controversial. T helper (Th) 17 T cells contribute to phagocytic function during upper airway colonisation while CD4+ T cells are necessary for optimal clearance of bacteria from the lung in the initial stages of pneumonia.22 ,23 In contrast, selective CD4+ T-cell depletion or non-selective T-cell inhibition reduces invasive disease29 but CD8+ T-cell depletion worsens pneumonia outcome.30 Our results suggest that FasL-dependent regulation of activated T cells could be a key factor in ensuring optimal pneumococcal clearance by pulmonary neutrophils early in the course of pneumonia and if this fails there is a more rapid progression to mortality. Since gld mice have relatively lower numbers of CD8+ T cells,27 and these cells can prominently express FasL, our results are consistent with an early role for these cells in pneumonia. Important roles for other T-cell populations such as CD4+ T-cells are equally possible in keeping with observations that T-cell depletion in fulminant disease is beneficial, since the concept of improving outcomes by reducing host responses in more fulminant disease are well established.21 However, alterations in T-cell activation may be crucial. The gld phenotype is associated with a large population of preactivated T cells, which leads to hypersecretion of Th1 cytokines and localised perturbation of cytokine networks could alter neutrophil function.31 Excess T-cell activation harms host defence; Th1-derived cytokines impair innate function when present at inappropriately high or sustained levels; interferon γ can inhibit Fcγ-mediated phagocytosis and intracellular killing of bacteria.32 ,33 Therefore excessive immune activation may have a paradoxical effect on innate responses to S pneumoniae in the lung and be as harmful as deficiency. Reducing T-cell activation during selective FasL deficiency or in fulminant disease can improve pneumonia outcome.29
Termination of immune responses requires FasL-induced T-cell apoptosis during sustained responses to micro-organisms.8 DcR3-a reduced T-cell activation and the defect in neutrophil-dependent bacterial clearance in gld mice, suggesting unopposed LIGHT/TL1A mediate the inappropriate T-cell activation in gld mice. Consistent with this possibility T-cell derived LIGHT/TL1A drive Th1 and Th17 cytokine expression at mucosal surfaces34 ,35 and favour dysregulated inflammatory responses.36 ,37 Although DcR3-a could alter neutrophil or macrophage responses directly the absence of evidence of macrophage or intrinsic neutrophil defects make this unlikely.
The benefits of FasL in our model are consistent with requirements for FasL to regulate T-cell activation during the early evolution of pulmonary neutrophil responses. The results of Matute-Bello et al suggest incomplete (lpr mice) or non-selective inhibition (DcR3-a) of Fas at a later stage of pneumococcal pneumonia improve outcomes.6 Reduction of neutrophil numbers or function, when these exceed the optimal amount required for bacterial clearance, can improve outcome21 ,38 and we believe the effects Matute-Bello et al observed are consistent with this requirement to decrease neutrophil responses. We found that lpr or DcR3-a-treated wild-type mice had no worse outcome than wild-type mice, while only the complete but selective inhibition of FasL in gld mice worsened outcome in all but the most advanced stages of pneumonia. It therefore seems that FasL signalling and that of related TNFSF ligands must be finely tuned to ensure optimal host responses during pneumonia (online supplementary figure 4).
Examination of clinical cohorts will be required to tease out these paradigms and confirm the therapeutic potential in CAP. Although lpr/gld mice are useful models of immune homeostasis and clinical diseases, as the murine phenotype varies considerably with genetic background, translation in clinical studies is essential.8 The potential translational implications are that manipulation of TNFSF ligands, an emerging therapeutic approach in oncology and other settings,39 could improve the outcome of CAP. Increased FasL expression has been documented in patients with pneumococcal infection.5 ,6 Documentation of relative decreases in FasL expression, altered ratios of DcR3 ligand expression leading to increased LIGHT/TL1A relative to FasL or enhanced T-cell activation in patients with worse clinical outcomes could trigger TNFSF ligand neutralisation using DcR-3 neutralisation or similar approaches early in pneumonia. This approach might also be beneficial when there is excessive neutrophilic inflammation later in disease. Our findings suggest that T-cell activation must be tightly controlled and confirm an important role for FasL expression in regulating host responses during pneumococcal pneumonia. These results also suggest modulation of DcR3 ligands could represent an unexploited therapeutic strategy that improves outcomes during CAP.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online materials 1
Footnotes
-
Funding This work was supported by a grant from the Sheffield Hospitals Charitable Trust Research Grant (No. 7843), a Wellcome Trust Senior Clinical Fellowship to DHD (No. 076945) and a British Lung Foundation fellowship to HMM (F05/7). SRW is a Wellcome Clinician Scientist (No. 078244) and AART an MRC Clinical Research Training Fellow (G0802255).
-
Competing interests DRW and VJW are employees and have stock in Eli Lilly & Company.
-
Provenance and peer review Not commissioned; externally peer reviewed.