Article Text

Abstract
Background Exposure to traffic-derived air pollutants, particularly diesel emissions, has been associated with adverse health effects, predominantly in individuals with pre-existing respiratory disease. Here the hypothesis that this heightened sensitivity reflects an augmentation of the transient inflammatory response previously reported in healthy adults exposed to diesel exhaust is examined.
Methods 32 subjects with asthma (mild to moderate severity) and 23 healthy controls were exposed in a double-blinded crossover control fashion to both filtered air and diesel exhaust (100 μg/m3 PM10) for 2 h. Airway inflammation was assessed by bronchoscopy 18 h postexposure. In addition, lung function, fraction of exhaled nitric oxide and bronchial reactivity to metacholine were examined in the subjects with asthma.
Results In healthy control subjects a significant increase in submucosal neutrophils (p=0.004) was observed following the diesel challenge. Significant increases in neutrophil numbers (p=0.01), and in the concentrations of interleukin 6 (p=0.03) and myeloperoxidase (p=0.04), were also seen in bronchial wash after diesel, relative to the control air challenge. No evidence of enhanced airway inflammation was observed in the subjects with asthma following the diesel exposure.
Conclusions Exposure to diesel exhaust at concentrations consistent with roadside levels elicited an acute and active neutrophilic inflammation in the airways of healthy subjects. This response was absent in subjects with asthma, as was evidence supporting a worsening of allergic airway inflammation.
- Inflammation
- air pollution
- asthma
- particulate matter
- diesel
- airway epithelium
- asthma mechanisms
Statistics from Altmetric.com
Introduction
Exposure to traffic-related pollution has been associated with retarded lung development,1 and enhanced allergic and respiratory symptoms in children living in close proximity to,2 or attending schools near, heavily trafficked roads.3 In addition, proximity to traffic or exposure to traffic-derived pollutants has been shown to exacerbate asthma symptoms in both children and adults.4 5 In the majority of these studies, in addition to traffic density, the greatest health impacts have been reported to be associated with roads carrying a high proportion of diesel-powered heavy and light goods vehicles.3 6 Consistent with these observational studies, diesel exhaust emissions have been shown to elicit pulmonary inflammation,7 increased airway resistance8 and alterations in vascular tone9 in controlled exposures of healthy subjects. In those with asthma with stable disease, on continuous treatment with inhaled corticosteroids, exposure to diesel exhaust has been associated with a significant increase in the degree of bronchial hyper-responsiveness determined 24 h after exposure.10
The potential toxicity of diesel emissions was recently highlighted in a London field study in which subjects with asthma were exposed to either a high diesel (Oxford Street) or a low traffic environment (Hyde Park).11 A persistent reduction in forced expiratory volume in 1 s (FEV1) occurred after the subjects walked along Oxford Street and signs of increased airway inflammation in the subjects with asthma were observed 24 h after exposure to the Oxford Street aerosol, based on the concentration of the neutrophil degranulation product myeloperoxidase (MPO) in induced sputum. The responses observed were attributed to diesel emissions, as Oxford Street is restricted to diesel-powered buses and taxis; however, the authors were careful to state that the exposure was more correctly viewed as being related to diesel traffic. While it is clear that diesel exhaust particulate matter (PM) is bioactive in vivo7–15 and in vitro,16–18 it should also be noted that the air-shed at roadside environments is comprised not only of primary tail pipe emissions, largely elemental and organic carbon, but also has contributions from brake wear, tyre wear, road surface abrasion and resuspension in the wake of passing traffic.19 These later non-exhaust sources, often characterised by elevated concentrations of transition metals,20 have been estimated to account for approximately half of the PM concentration observed at the roadside,21–23 and often predominate in countries with significant road sanding and studded tyre use during the winter months.24 25 The potential contribution of these components to the observed health effects is sometimes ignored at a regulatory level, which focuses on the reduction of tail pipe emissions through improvements in engine technology.
To define the actual contribution of diesel exhaust emissions to responses of subjects with asthma in the real world, we performed controlled exposures of healthy subjects and subjects with asthma to freshly generated diesel exhaust (100 μg/m3 PM10, median aerodynamic diameter 80 nm, characterised as a largely organic aerosol12) and filtered air. We have previously demonstrated acute airway neutrophilic inflammation in healthy subjects exposed to diesel exhaust at this exposure dose12 and hypothesised that this response would be augmented in those with asthma. In addition, we examined the induction of allergic airway inflammation in response to diesel PM, as an alternative mechanism for their symptomatic responses to environmental PM. Neutrophilic and allergic inflammation was assessed using a comprehensive series of markers reflecting recruitment (proinflammatory mediators and adhesion molecules), tissue and airway cell numbers, as well as markers of cellular activation. Lung function (dynamic spirometry and peak expiratory flow), exhaled nitric oxide (NO) and bronchial hyper-responsiveness to metacholine were also examined in the subjects with asthma.
Methods
Subjects
Healthy subjects and subjects with asthma were invited to participate in the present study through advertisements. They had to fulfil the following inclusion criteria: aged between 18 and 45 years, never smokers, with normal lung function (FEV1 and forced vital capacity (FVC) >80% of predicted, normal FEV1/FVC ratio), plus the absence of concomitant diseases, apart from allergy in the asthmatic group. Two groups of subjects with asthma were recruited based on asthma severity according to GINA guidelines (Global Initiative for Asthma, http://www.ginasthma.com): those with mild asthma on β2-agonist treatment on demand only, and those with moderate asthma demanding inhaled corticosteroid treatment. All individuals with asthma had to have a positive history of allergy and at least one positive skin prick test against a standard panel of common aeroallergens. In those with mild asthma, bronchial hyper-responsiveness (PC20 <8 mg/ml metacholine) was demanded. Exclusion criteria were uncontrolled asthma, airway infection within 6 weeks prior to, or during the study, and current use of medication other than inhaled corticosteroids and short-term β2-agonists as outlined above. During the study, the subjects were not allowed to take any additional medication or antioxidant supplements. The study was performed with the approval of the local research ethics committee, in accordance with the Declaration of Helsinki, and with the written informed consent of all participants.
Study design
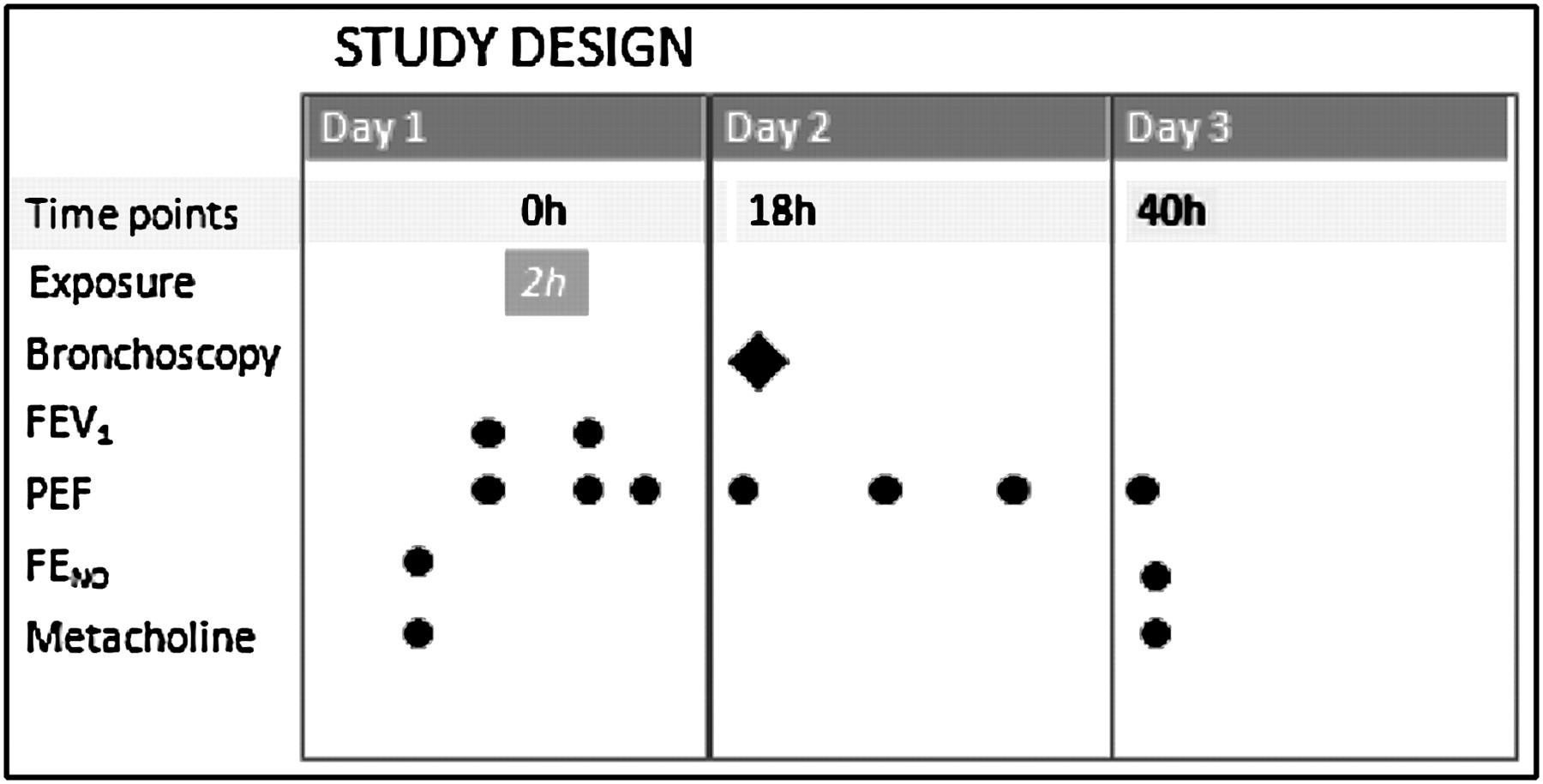
The study was carried out outside the pollen season. Using a randomised, double-blind, crossover study design, each subject was exposed in an exposure chamber on two occasions, at least 3 weeks apart, as previously described (figure 1).11 All subjects were exposed to dilute diesel exhaust at a PM concentration of 100 μg/m3 and filtered air, thus acting as their own controls. The exposures lasted for 2 h, during which subjects alternated 15 min intervals of exercise on a bicycle ergometer with 15 min of rest. For each subject, the ergometer workload was calibrated to achieve a ventilation of 20 l/m2 of body surface, to ensure a similar exposure on both occasions. During the 2 h of exposure, symptom scores were recorded every 30 min. Bronchoscopy with biopsy sampling and airway lavages was performed 18 h after the end of each exposure, as previously described.7 8

Study design. Subjects were exposed in an exposure chamber in a double-blinded fashion to both filtered air and diesel exhaust (100 μg/m3 PM10) for 2 h. Airway inflammation was assessed by bronchoscopy 18 h postexposure. In the subjects with asthma, lung function was assessed before and immediately after the exposures, along with peak expiratory flow (PEF) measurements before, immediately after exposure and repeatedly until the morning of day 3. Measurements of exhaled nitric oxide (NO) were performed at inclusion and 18 h after exposure. Metacholine challenges were assessed at inclusion and 40 h postexposure. FENO, fraction of exhaled nitric oxide; FEV1, forced expiratory volume in 1 s.
In the subjects with asthma, lung function was assessed before and immediately after exposure, along with peak expiratory flow measurements immediately before exposure, after exposure and repeatedly until the morning of the day after exposure. Measurements of exhaled NO were carried out at inclusion and 18 h postexposure. Metacholine challenges were performed at inclusion and 40 h postexposure (figure 1).
Diesel exhaust exposure
For consistency with our previous studies, diesel exhaust was generated from an idling 1991 Volvo diesel engine (Volvo TD45, 4.5 l, four cylinders, 680 rpm), as previously described.7 The steady-state concentration of PM, gases and semi-volatiles during the diesel exposures were: for the healthy group 96 (7) μg/m3 (PM10), 9.4 (2.2) ppm (CO), 1.3 (0.11) ppm (NO), 0.41 (0.03) ppm (nitrogen dioxide), 1.7 (0.12) ppm (oxides of nitrogen) and 1.2 (0.44) ppm (total gaseous hydrocarbons-C3H8-equivalent) and for the asthmatic group 97 (11) μg/m3 (PM10), 7.8 (2.6) ppm (CO), 1.2 (0.06) ppm (NO), 0.36 (0.05) ppm (nitrogen dioxide), 1.6 (0.53) ppm (oxides of nitrogen) and 1.0 (1.1) ppm (total gaseous hydrocarbons-C3H8-equivalent), expressed as mean and SDs. More than 90% of the exhaust was shunted away, and the remainder was diluted with filtered air heated to 20°C (relative humidity ∼50%) before being fed into a whole-body exposure chamber (3.0×3.0×2.4 m) at a steady-state concentration. The chamber was monitored continuously for pollutants, with exposures standardised with the use of the NO concentration to deliver a PM concentration of 100 μg/m3. The PM mass in the exposure chamber was dominated by fine particles (<1 μm) in the accumulation mode, with a mass median particle diameter for the submicrometre sized PM of 0.18 μm.
Bronchoscopy and processing of samples
Bronchoscopy was performed 18 h postexposure using a flexible video bronchoscope (Olympus BF IT200, Tokyo, Japan), as previously described.7 8 This time point was selected as based on previous studies indicating a peak inflammatory response at this time point. Prior to bronchoscopy, the subjects with asthma inhaled 0.2 mg of salbutamol dry powder. Bronchial biopsies were taken either from the anterior aspect of the main carina and the subcarinae of the third and fourth generation airways of the right side or from the posterior aspect of the main carina and the corresponding subcarinae on the left side. Bronchial wash (BW, 2×20 ml) and bronchoalveolar lavage (BAL, 3×60 ml) were carried out on the contralateral side, in a predetermined randomised way. The aspirates recovered from the 20 ml instillations of the BW as well as the BAL recovery were collected into separate siliconised containers and immediately placed on ice. All lavage samples were filtered through nylon (pore diameter 100 μm) and centrifuged at 400 g for 15 min. The supernatants were separated from the cell pellet and analysed for interleukin 6 (IL-6), IL-8, MPO and stem cell factor (SCF). IL-6, IL-8 and SCF were measured using commercially available ELISA kits (R&D Systems, Minneapolis Minnesota, USA). MPO was analysed using an MPO radioimmunoassay (Pharmacia AB, Uppsala, Sweden). Cell pellets were re-suspended in phosphate-buffered saline (PBS) at a cell concentration of 106 cells/ml. Differential cell counts were performed on cytocentrifuge preparations stained with May–Grünwald Giemsa, and 400 cells per slide were counted.
Immunohistochemistry
Endobronchial mucosal biopsies were processed into glycolmethacrylate resin, as previously described.7 Sections of 2 μm thickness were cut and stained immunohistochemically using the streptavidin–biotin–peroxidase technique with monoclonal antibodies (mAbs) directed against specific cellular markers to detect inflammatory cells in the bronchial mucosa. Stained inflammatory cells (neutrophils, mast cells, eosinophils, CD3+, CD4+, CD8+ lymphocytes, CD25+ and CD68+ cells) were counted in the epithelium and in the submucosa excluding glands, blood vessels and muscle. The counts were expressed as cells/mm in the epithelium and cells/mm2 in the submucosa, and counted using a light microscope. The length of the epithelium and the area of the submucosa were calculated using a computer-assisted image analyser (Zeiss KS400 software, Image Associates, Bicester, UK). Vascular endothelial adhesion molecules (P-selectin, E-selectin, vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1)) in the vessels were quantified by expressing the number of vessels stained with specific mAbs as a percentage of the total number of blood vessels stained with the pan-endothelial mAb EN4 in adjacent 2 μm sections.
Lung function tests
Standard lung function tests including FVC and FEV1 were performed at inclusion, using a spirometer (Vitalograph R, Buckingham, UK). At least three satisfactorily performed and well-cooperated measurements of each variable were performed as judged by an experienced lung function technician and according to the guidelines of the American Thoracic Society.26 In addition, in the asthmatic group, lung function tests were performed immediately before and immediately after the exposures. The subjects with asthma were also equipped with a peak expiratory flow (PEF) metre (PICO 3; Ferraris Medical, Hertford, UK) and PEF measurements were performed immediately before (0 h) each exposure, immediately after (2 h), the evening of day 1 (8 h), the morning of day 2 (18 h), the afternoon of day 2 (24 h), the evening of day 2 (32 h) and the morning of day 3 (42 h), after both the filtered air and diesel exhaust exposures.
Fraction of exhaled nitric oxide (FENO)
FENO at a flow rate of 50 ml/s was assessed in the asthmatic group. Measurements were carried out 18 h after each exposure, using a chemiluminescence analyser (NiOX; Aerocrine AB, Stockholm, Sweden).
Metacholine challenges
Measurements of airway responsiveness were performed in the asthmatic group 3–7 days before each exposure and 40 h after exposure. Metacholine challenges were performed according to the method described by Juniper et al.27 The test aerosols were generated continuously by a Wright's nebuliser (Roxon Medi-Tech, Montreal, PQ, Canada), delivered into a Hans Rudolph two-way valve (type 1410B, Somedic sales AB, Farsta, Sweden) and inhaled through a mouthpiece by quiet tidal breathing for 2 min with the nose clipped. The nebuliser output was 0.13 ml/min. Initially, saline was inhaled, followed by doubling concentrations of metacholine (0.06–32 mg/ml) at intervals of 5 min. The response was measured by FEV1 before and at 30, 90 and 180 s after each inhalation. Inhalations were discontinued when a fall in FEV1 of ≥20% below the lowest postsaline value was noted. Results were expressed as PC20 obtained from the log dose–response curve by linear interpolation of the two last points expressed in non-cumulative units.
Statistical analysis
The present study was powered using BW neutrophilia as the primary end point using an SD in the response variable of 0.83 based on our previous observations.12 This indicated that we required a minimum group size of 16 to detect a treatment difference at a two-sided 0.05 significance level and a probability of 80%, if the true difference between treatments was 1.280×104 cells.12
Lung function responses were normally distributed, as established using the Shapiro–Wilks normality test, and are therefore reported as means with SD. Measurements in the bronchial lavages and biopsies were not normally distributed, assessed as outlined above, and are therefore reported as median values with the 25th and 75th percentiles. All paired air versus diesel exhaust comparisons were performed using the Wilcoxon signed ranks test. Pseudo-baseline comparisons, after air challenge between groups, were performed using the Mann–Whitney U test. Correlations between diesel-induced responses were performed using the Spearman rank order correlation. For analyses of the impact of the diesel challenge on serial PEF measurements, a linear mixed-effects model was employed, with subject included as a random effect, and period, time and exposure as fixed effects. The exposure×time interaction was included in the model to determine whether changes in outcome over time were different after diesel exhaust, as compared with air exposure. A heterogeneous first-order autoregressive covariance structure was used to model the correlation between repeated observations. Overall PEF responses after air and diesel exhaust were summarised as the area under the curve and compared using the Student paired t test. All statistical analyses were performed using SPSS, version 15.0 (SPSS, Cary, North Carolina, USA). A p value of <0.05 was considered statistically significant.
Results
Twenty-three healthy non-atopic subjects and 32 atopic persons with controlled mild to moderate asthma, according to GINA guidelines (http://www.ginasthma.com), fulfilled the inclusion criteria for this study. Subject demographics are presented in table 1. Sixteen subjects with mild asthma, hyper-reactive to metacholine (PC20 <8 mg/ml) and treated with only short-acting inhaled β2-agonists on demand were included. Additionally, 16 subjects with moderate asthma, treated with inhaled corticosteroids (200–1200 μg/day of budesonide or equivalent), out of whom five were hyper-responsive to metacholine, also participated. All subjects tolerated the diesel exhaust exposure well and reported no symptoms of respiratory problems during the exposures or in the subsequent 40 h. BW recoveries were 33 and 31% in healthy subjects, 30 and 29% in those with mild asthma, and 29 and 30% in those with moderate asthma, following air and diesel exhaust exposure, respectively. Recoveries in BAL were 72 and 70% in healthy subjects, 71 and 68% in those with mild asthma, and 73 and 74% in those with moderate asthma, following air and diesel exhaust exposures.
Subject demographics
Neutrophilic inflammation
In healthy subjects, exposure to diesel exhaust induced a neutrophilic inflammation in the airways detected both in the BW wash (p=0.01) and in the bronchial submucosa (p=0.004), compared with the air exposure (figure 2). There was also an increase in BW IL-6 (p=0.03) and BW MPO (p=0.04) following the diesel exhaust challenge (figure 2). No significant changes were detected in the levels of IL-8, or in the expression of the endothelial adhesion molecules ICAM-1, P-selectin or E-selectin. The increases in IL-6 and MPO at 18 h postexposure were associated with the increase in BW polymorphonuclear leucocyte numbers (r=0.50, p=0.02; and r=0.54, p=0.007, respectively), but not with the neutrophilic response in the submucosa. No significant changes in BAL fluid or bronchial epithelial markers of inflammation were observed in the healthy subects or subjects with asthma following the diesel challenge (data not shown). In the total and individual asthmatic groups, no significant diesel exhaust-induced changes in BW or BAL neutrophils, IL-6, IL-8, MPO or submucosal neutrophils and ICAM-1 were detected (figure 1).

Following the ‘neutrophilic pathway’; responses after air and diesel exhaust (DE) exposure. The first two bars represent responses in the healthy control group and bars three and four represent responses in subjects with asthma. Bars five and six represent subjects with mild asthma, and bars seven and eight responses in subjects with moderate asthma. Healthy subjects responded to DE exposure with a neutrophilic inflammation; this response is not detected in those with asthma. Data are given as medians with IQR; whiskers denote minimum and maximum values. BW, bronchial wash; ICAM-1, intercellular adhesion molecule 1.
Allergic inflammation
Allergic inflammation (eosinophils, mast cells in BW and bronchial submucosa vascular endothelial adhesion molecules (VCAM-1 and SCF) was assessed in all subjects. As expected, there was a difference at baseline between healthy subjects and those with asthma, with greater numbers of mast cells and eosinophils in the asthmatic group (table 2). There were no baseline differences in inflammatory cells in the BAL (data not shown), except from significantly greater concentrations of MPO in the asthmatic group: healthy subjects 0.00 (0.00–1.45) and subjects with asthma 1.46 (0.00–4.98) μg/l; p=0.02. Among markers for allergic inflammation, the only significant diesel exhaust-induced change was found in the healthy group, with an increase in submucosal mast cells: 19 (11–27) cells/mm2 after air versus 21 (16–35) after diesel exhaust; p=0.004 (figure 3).
Differences between healthy control and allergic asthmatic groups after air exposure
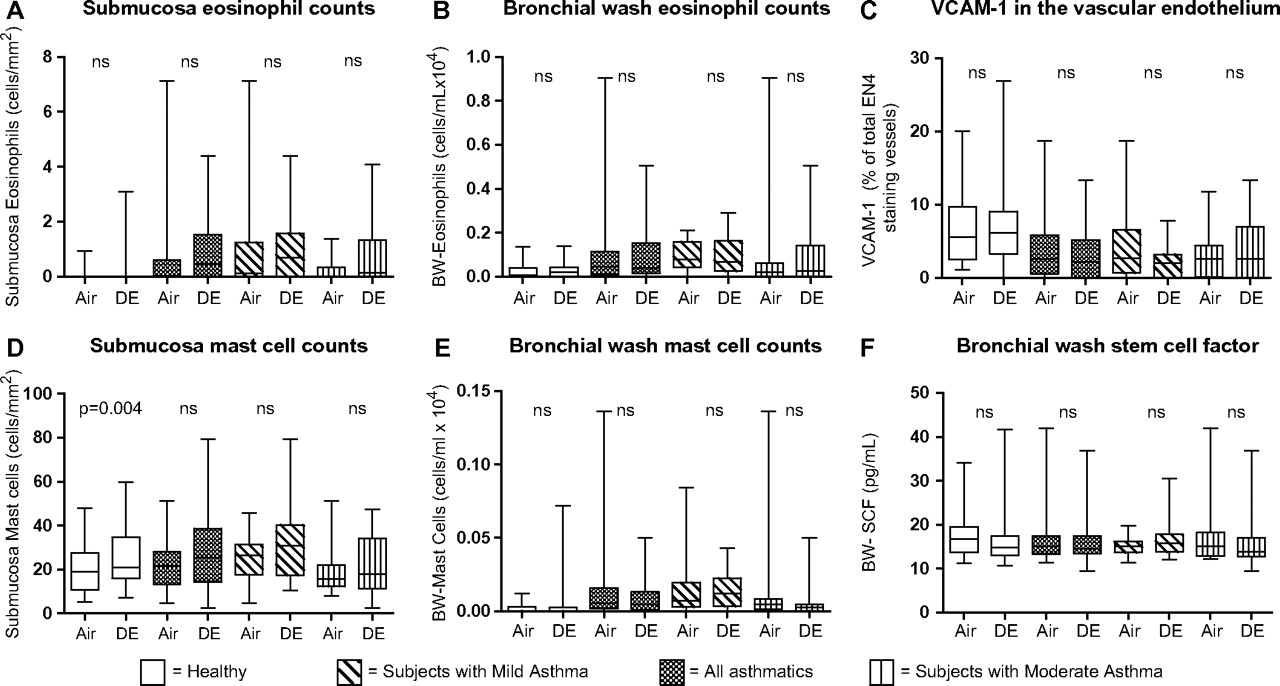
Following the ‘allergic pathway’; responses after air and diesel exhaust (DE) exposure. The two first bars represent responses in the healthy control group and bars three and four represent responses in subjects with asthma. Bars five and six represent subjects with mild asthma, and bars seven and eight responses in subjects with moderate asthma. Data are given as medians with IQR; whiskers denote minimum and maximum values. BW, bronchial wash; SCF, stem cell factor; VCAM-1, vascular cell adhesion molecule 1.
Lung function responses
Lung function measurements were only assessed in the asthmatic group. A small but statistically significant decrease in FEV1 immediately after the diesel exhaust exposure (before 4.03 (0.79); after 3.98 (0.79); p=0.01) was observed. However, when comparing the prechange and postchange, between air and diesel exposures, this response did not differ between groups (p=0.30). Controlled exposure to diesel exhaust appeared to induce a reduction in PEF which failed to reach statistical significance (figure 4). In all those with asthma, PEF values were decreased by 8% at 18 h and 14% at 24 h after diesel exhaust relative to pre-exposure values, with little difference between the moderate and mild subgroups (figure 4B–C). Overall when the PEF responses were summarised as the area under the curve over the 42 h period, no statistically significant differences were observed in either asthmatic group following diesel challenge.
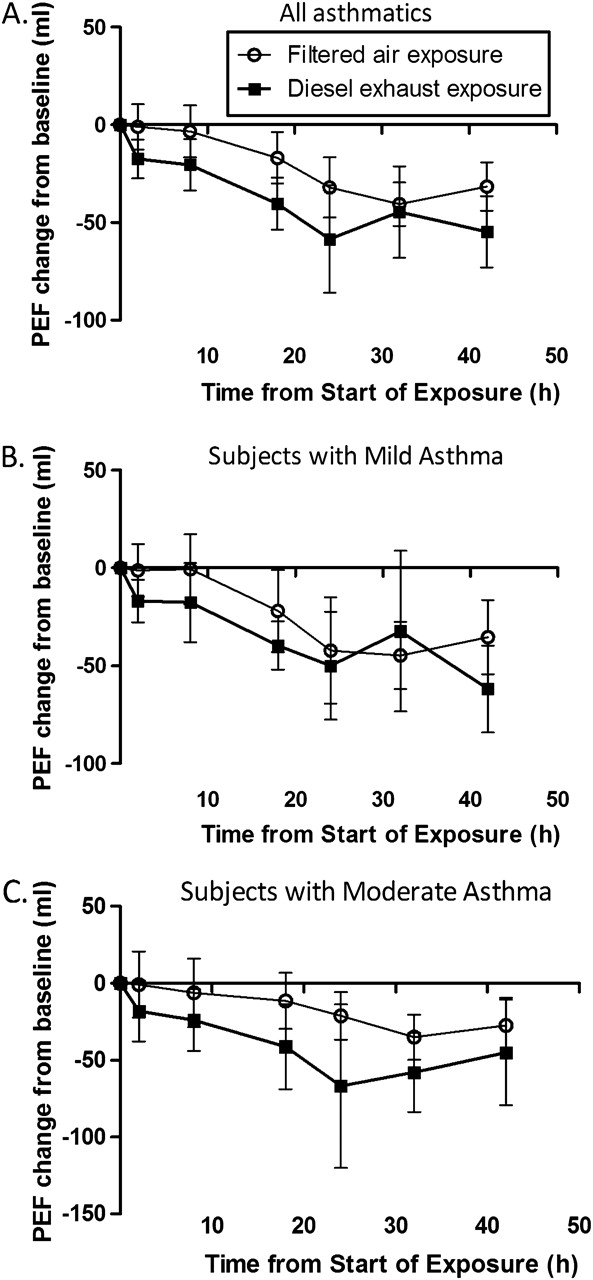
Mean change in peak expiratory flow (PEF) before and repeatedly after a controlled exposure to filtered air and diluted diesel exhaust (100 μg/m3 for 2 h). Values are given as absolute change from initial value for all participants in A, those with mild asthma in B and those with moderate asthma in C. Data are given as means; І bars represent the 95% CI.
FENO and metacholine responses
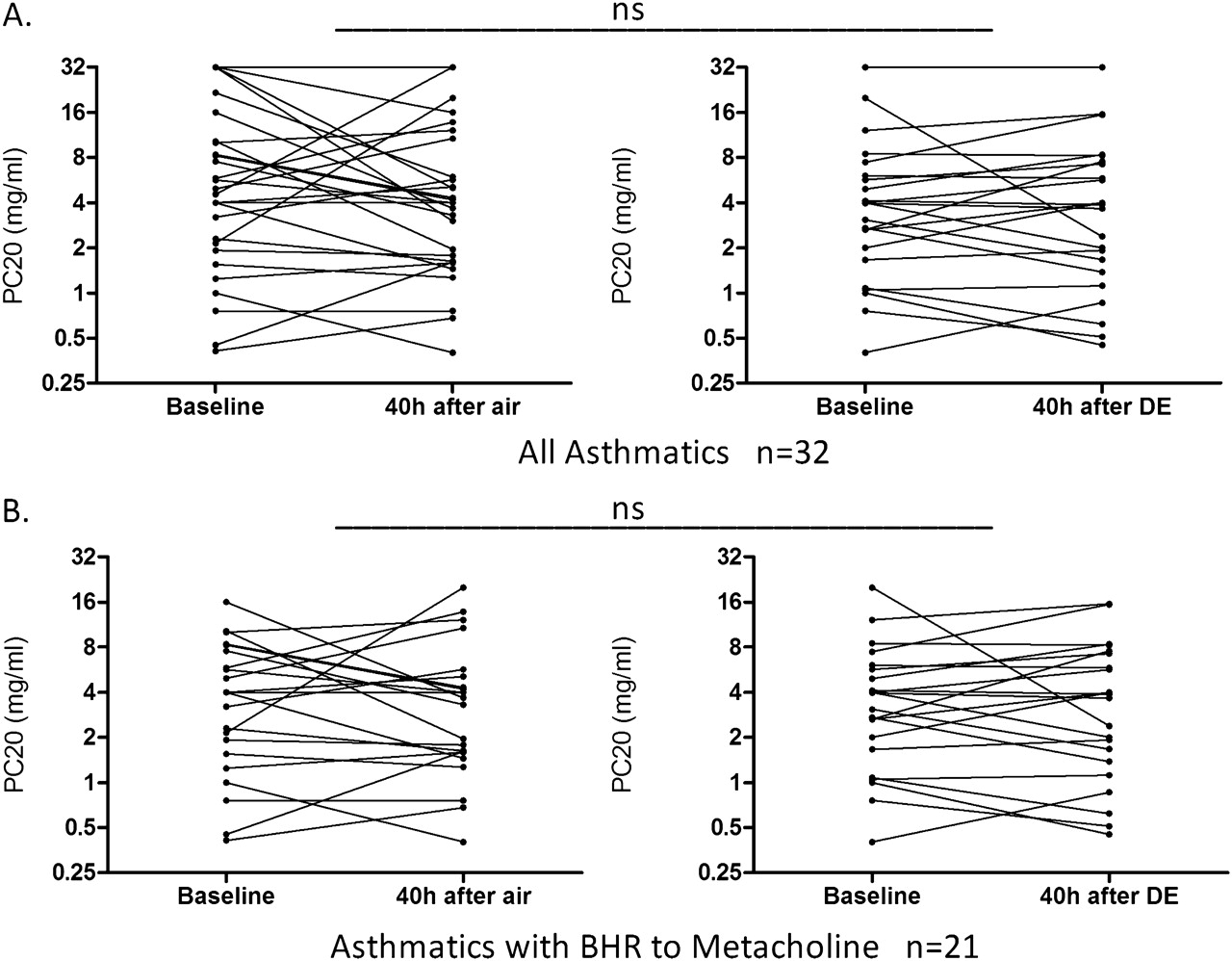
FENO and metacholine challenges were only performed in the asthmatic group. There were no significant differences between the changes in FENO following exposure to filtered air and diesel exhaust. Neither was a significant change in FENO found when those with asthma were divided into mild and moderate subgroups (figure 5). Exposure to diesel exhaust did not increase bronchial hyper-responsiveness in the whole group of subjects with asthma at 40 h postexposure (figure 6A). A subgroup analysis was also performed, evaluating only those with asthma that were hyper-responsive at study inclusion, but no change was found in hyper-responsiveness following diesel exhaust compared with filtered air exposure (figure 6B).

Fraction of exhaled NO (FE-NO, at a flow rate of 50 ml/s) 18 h after exposure to filtered air and diesel exhaust (DE) in subjects with mild and moderate asthma. Data are given as medians with IQR; whiskers denote minimum and maximum values. No significant changes were detected following DE exposure (Wilcoxon signed ranks test).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Metacholine provocative concentration causing a 20% fall in forced expiratory volume in 1 s (PC20) in 32 subjects with asthma at 40 h following exposure to air and diesel exhaust (DE). Please note that the y-axes indicate doubling concentrations. A includes all 32 subjects with asthma. B includes only subjects with asthma with bronchial hyper-responsiveness (BHR) to metacholine at inclusion, defined as PC20 <8 mg/ml. Comparisons in PC20 were performed between the air and DE challenge using the change in this parameter at 40 h relative to baseline.
Discussion
The recent US Health Effects Institute special report examining the evidence linking traffic emissions to health effects concluded that the available (largely epidemiological) data were sufficient to suggest a causal link with asthma exacerbations.28 The report also highlighted, together with other recent reviews and editorials,29 30 uncertainties and gaps in our current knowledge concerning the causative components and mechanisms driving these effects in individuals exposed to the roadside air-shed. In the present study we examined the hypothesis that diesel exhaust (PM10, plus associated gaseous emissions) would elicit an augmented inflammatory response in subjects with asthma, consistent with the increased symptoms observed in individuals living or working near busy roads.2–6 To investigate this, we employed a freshly generated diesel exhaust aerosol (previously characterised12), from an idling engine, as a surrogate for roadside exposures in high diesel microenvironments.
Healthy volunteers and subjects with asthma were exposed to dilute diesel exhaust at a PM10 concentration of 100 μg/m3, a level consistent with that observed at kerbside locations in London (http://www.londonair.org.uk/), and comparable with the concentrations reported in the recent Oxford Street field study, in which upper airway inflammation and persistent lung function decrements were observed in subjects with asthma walking along a road carrying only diesel-powered vehicles.11 Previous work has also demonstrated airway inflammation in healthy subjects 6–24 h after exposure to experimental diesel exhaust challenges,7 8 12 13 with some evidence of increased airway resistance.10 In addition, in high dose exposure studies (PM10 300 μg/m3) an upregulation of redox-sensitive signalling pathways has been observed in healthy subjects,31 consistent with inhaled diesel exhaust particles inducing inflammation through the imposition of oxidative stress at the air–lung interface. In contrast, while a clinically significant increase in bronchial hyper-responsiveness has been reported in subjects with asthma treated with inhaled corticosteroids after exposure to a similar diesel exhaust concentration, no enhanced airway inflammatory changes were found in induced sputum in this group.10
In the present study, we again confirmed the recruitment and activation of neutrophils in the airway lumen and bronchial mucosa of healthy subjects following a diesel exhaust challenge consistent with urban concentrations, but found no evidence of an equivalent, or augmented response, in either the mild or moderate asthmatic groups. Furthermore, airway eosinophil and mast cell numbers were unaltered following exposure to diesel exhaust. As multiple markers of the induction of neutrophilic and allergic inflammation were assessed encompassing ‘early’ markers of cellular recruitment to ‘late’ markers of activation within the airway lumen, we contend that the acute inflammatory response appeared absent rather than simply delayed. It should be noted that previous investigations of healthy controls and those with mild asthma exposed to diesel exhaust, with responses evaluated at 6 h, also failed to demonstrate enhanced inflammation in the latter group.8
There were no significant changes in PEF values following diesel exhaust in either those with mild or moderate asthma. However, as shown in figure 4, considerable heterogeneity was apparent between subjects, with PEF reductions of >100 ml in certain individuals, representing a clinically significant bronchoconstrictive response. Previously we have reported an increase in bronchial hyper-reactivity in subjects with asthma treated with inhaled corticosteroids 24 h following a high dose diesel exhaust exposure (300 μg/m3).10 Since bronchoscopy was performed at 18 h postexposure, when the airway inflammation was expected to peak, metacholine challenge was carried out the following day—that is, 40 h after the end of exposure. As a comparable response was not seen in the current investigation after the lower PM concentration of 100 μg/m3, this may imply either a dose relationship or that the bronchial hyper-reactivity had peaked earlier.
In the recent Oxford Street study, subjects with asthma responded with increased bronchoconstriction (decreased FEV1) along with signs of airway inflammation, as determined in induced sputum, when exposed to traffic (diesel)-derived PM.11 In the present study, FEV1 was only measured before and immediately after exposures. Thereafter lung function responses were determined using PEF metres. A significant bronchoconstrictive response was not detected up to 24 h postexposure in those with asthma, irrespective of disease severity or medication. While this may reflect differences in the asthma phenotypes between the two studies, it may also reflect differences in the causative triggers of the response between a controlled tail pipe emission and the real-world roadside aerosol. The precise composition and size distribution of the diesel aerosol at the roadside will depend on a number of factors, including fuel composition, drive cycle and engine technology.17 The controlled exposure in the current study can therefore only be viewed as a generalised diesel exposure, reflecting an idling scenario and producing an aerosol of ultrafine organic-rich particles.12 Of major importance, the absence of airway inflammation in the subjects with asthma in this study is consistent with other controlled chamber studies performed using either concentrated ambient particles which were delivered to subjects at high traffic locations (2 h exposures to ∼200 μg/m3 PM<2.5 μm)32 or experimentally generated ultrafine carbon particles (2 h exposures to 10 μg/m3, average diameter 25 nm), as a surrogate for elemental carbon diesel emissions.33
In epidemiological studies it has been difficult to distinguish diesel exhaust-related effects from those of other components of traffic mixture. The ambient particulate air-shed at roadside locations is complex, consisting of both primary tail pipe emissions and fugitive emissions related to vehicular wear processes and the resuspension of road dust.18–24 This complexity highlights the valuable role of controlled chamber exposures in complementing real-world studies, by permitting components of the aerosol to be examined in isolation, in this case fresh tail pipe emissions. It is of major importance to explore the role of different sources and causative agents in the traffic pollution mixture further, since this will increase our ability to protect the health of people with asthma.29 34 So far, current regulatory strategies to reduce PM-induced health effects have mainly targeted tail pipe emissions through improved engine, fuel and exhaust technologies.
It is now well established that exposure to diesel exhaust at concentrations consistent with roadside levels elicits an acute neutrophilic inflammation in the airways of healthy subjects. The present study, as well as preceding investigations, did not demonstrate any acute airway inflammatory response following exposure to diesel exhaust. This suggests that the increased sensitivity in subjects with asthma to traffic-related air pollution is not necessarily associated with a classical acute inflammation or aggravation of standard cellular indicators of allergic asthmatic inflammation. The mechanisms underlying the differential airway responses to diesel exhaust between healthy subjects and those with asthma, as well as the potential role of coarse PM components in the traffic air-shed, demand further investigation.
Acknowledgments
The authors would like to thank Annika Johansson, Helena Tjällgren, Helen Bertilsson, Jamshid Pourazar, Ann-Britt Lundström and Maj-Cari Ledin for their technical contribution to the project. We also would like to express our gratitude to the volunteers who participated in this investigation.
References
Footnotes
See Editorial, p 4
Linked articles 145391.
Funding Swedish Heart–Lung Foundation, European Commission HEPMEAP project (QLRT 1999-01582) and Umeå University, Sweden. AB is the holder of the Lars Werkö distinguished research fellowship from the Swedish Heart–Lung Foundation.
Competing interests None.
Ethics approval This study was conducted with the approval of the research ethics committee at Umea University, Sweden.
Provenance and peer review Not commissioned; externally peer reviewed.