Article Text

Abstract
Background: Malignant mesothelioma is a cancer which is refractory to current treatments. Imatinib mesylate is a selective inhibitor of tyrosine kinases such as bcr-abl, c-Kit, c-Fms and platelet derived growth factor receptor β (PDGFRβ). PDGFRβ is often overexpressed in mesothelioma cells and is a therapeutic target for imatinib in some solid tumours. A study was undertaken to assess whether imatinib alone or combined with chemotherapeutic agents may be effective for treating mesothelioma.
Methods: Cultures from mesothelioma MMP, REN and ISTMES2 cell lines were treated with imatinib alone or in combination with a chemotherapeutic agent.
Results: Imatinib induced cytotoxicity and apoptosis selectively on PDGFRβ positive mesothelioma cells via blockade of receptor phosphorylation and interference with the Akt pathway. Of the chemotherapeutic agents tested in combination with imatinib, a synergistic effect was obtained with gemcitabine and pemetrexed.
Conclusions: This study provides a rationale for a novel translational approach to the treatment of mesothelioma which relies on enhancement of tumour chemosensitivity by inhibition of Akt.
- HGF, hepatocyte growth factor
- HMC, human mesothelial cells
- LC50, lethal concentration killing 50% of cells
- M-CSF, macrophage colony stimulating factor
- MMe, malignant mesothelioma
- PDGF, platelet derived growth factor
- PDGFRβ, PDGF receptor β
- VEGF, vascular endothelial growth factor
Statistics from Altmetric.com
- HGF, hepatocyte growth factor
- HMC, human mesothelial cells
- LC50, lethal concentration killing 50% of cells
- M-CSF, macrophage colony stimulating factor
- MMe, malignant mesothelioma
- PDGF, platelet derived growth factor
- PDGFRβ, PDGF receptor β
- VEGF, vascular endothelial growth factor
Malignant mesothelioma (MMe) is an asbestos-related tumour, the incidence of which is expected to rise dramatically in Europe.1 In the USA the incidence of MMe has already increased by 90% in the last few years.2 Because of its biological aggressiveness, MMe is nearly always fatal except in rare less advanced cases, with a median survival of 12.6 months.3
A number of growth factors such as hepatocyte growth factor (HGF),4,5 vascular endothelial growth factor (VEGF)6,7 and insulin-like growth factor-1 and -28 have been shown to play a significant role in the development and progression of MMe. Moreover, several studies have reported a crucial role for platelet derived growth factor (PDGF) A and B in MMe cell growth.9 A high level of expression of PDGF receptor β (PDGFRβ) was seen in MMe cells but not in normal human mesothelial cells (HMC), mostly expressing PDGFRα.10 Furthermore, increased expression of PDGF A and B was detected at higher levels in MMe cells than in HMC,11 and a significant reduction in MMe cell growth or migration was observed by blocking PDGF A and B.12 Expression of c-Kit on MMe cells has been demonstrated by some authors, although its role in this tumour is controversial.13–15 Production of macrophage colony stimulating factor (M-CSF) by mesothelial cells has already been reported,16 and inhibition of c-Fms receptor by imatinib has been demonstrated.17
Many cytokines are released in the microenvironment by tumour stromal cells, and PDGF paracrine stimulation has been demonstrated in human tumours, particularly MMe.18,19 PDGFRβ activated by PDGF B can induce PI3K/Akt signalling20 which seems to be crucial for the survival of MMe cells.21
Imatinib is a selective inhibitor for a subset of tyrosine kinases including bcr-abl, c-Kit, PDGFRβ22 and c-Fms.17 PDGF receptors are expressed by several tumour cells and have been identified as potential therapeutic targets for imatinib.23 In mesothelioma the extent of PDGFRβ positive specimens ranges from about 30% to 45% in different studies.24,25 Although the therapeutic inefficacy of imatinib monotherapy for mesothelioma has recently been reported,25,26 combination therapies with imatinib in mice yielded successful results.27,28 Gemcitabine, cisplatin, etoposide, doxorubicin and, more recently, pemetrexed have been shown to be active in the treatment of MMe. Combined treatment with cisplatin/pemetrexed and cisplatin/gemcitabine have been found to be more effective than each single agent used alone.29 The aim of the present study is to investigate a translational approach which assesses the possible efficacy of imatinib as a single agent and in combination treatment for MMe.
METHODS
Cell cultures
Mesothelioma cells were derived from pleural effusions and stabilised in culture as continuous cell lines. MMP cells and primary HMC were characterised and cultured as previously described.5 REN cells were kindly provided by Dr Albelda and ISTMES2 were from the IST cell depository of Genoa (Italy).
Drugs
Imatinib was kindly provided by Novartis (Basel, Switzerland), and gemcitabine and pemetrexed were provided by Lilly (Indianapolis, Indiana, USA). Commercially available cisplatin, doxorubicin and etoposide were obtained from Alexis (Lausen, Switzerland).
Signal transduction
Cells were grown in 0.2% fetal bovine serum (FBS) for 24 h, then pre-incubated for 90 min in the presence or absence of 10 μM imatinib. Purified PDGF (R&D, Milan, Italy), 20 ng/ml, was added to the same medium. Immunoprecipitation and immunoblotting were performed as previously described.5 Antibodies used were: PDGFRβ, phospho-PDGFRβ, c-Kit, c-Fms (Santa Cruz Biotechnology, USA), phospho-Akt-Ser473 (Cell Signaling, USA), phosphotyrosine (UBI, USA) and phospho-Erk1/2 (Sigma, USA). Reactions were detected by the Enhanced Chemiluminescence System (ECL, Amersham, UK).
Cytotoxicity and apoptosis
Subconfluent cells in 96-well plates were exposed for 48 h to medium supplemented with 2% FBS, with or without different drugs at concentrations ranging from 1×10−10 M to 1×10−3 M. Cell viability was assessed by MTT assay30 on eight replicas at each concentration point to determine single drug lethal concentration (LC50) values. Normalised cytotoxicity percentages were obtained from the formula: (A570 mean values of extracts from treated samples/A570 mean values of extracts from untreated control samples) ×100.
LC50 values, calculated using Origin software (Microcal Software, USA), were used to draw the theoretical addictivity isobole according to the 50% isobologram method.31 A series of dose-response curves were then generated for each chemotherapeutic drug as above in the presence of several fixed concentrations of imatinib. The resulting LC50 values were plotted on the isobologram for assessment of the hypothetical superadditive effect.
Apoptosis was evaluated by TUNEL analysis (DeadEnd Colorimetric TUNEL System, Promega, USA) following treatment with imatinib, alone or combined with gemcitabine or pemetrexed, and the specific LC50 values were determined by MTT analysis in each cell type as follows. MMP: imatinib 3×10−7 M, gemcitabine 5×10−7 M, pemetrexed 6.5×10−6 M; REN: imatinib 1×10−6 M, gemcitabine 5×10−9 M, pemetrexed 1×10−5 M; ISTMES2: imatinib 4×10−6 M, gemcitabine 1×10−9 M, pemetrexed 5×10−6 M. In brief, subconfluent cells plated on glass slide flaskets (NUNC, Rochester, NY, USA) were exposed to medium supplemented with 2% FBS containing the different drugs for 48 h and subsequently fixed in 10% formalin. Biotin-dU positive nuclei were counted on 10 fields with at least 100 cells in the same slide. Values are expressed as the mean (±SE) percentage of positive nuclei of the total counted.
Statistical analysis
For the cytotoxicity assay we performed three separate experiments for each drug and drug combination in the different cell types. Data from each experiment are expressed as mean (SE) values of eight determinations for every concentration point. All mean values from each of the three experiments were used to calculate the curve with the best fit using the Origin software and to calculate the corresponding LC50 value with confidence limits by regression analysis. These LC50 values were compared using the Student’s t test with theoretically additive doses and their confidence intervals were calculated as described by Tallarida.32
For apoptosis, statistical differences between the theoretical additive effects of the chemotherapeutic agents (gemcitabine or pemetrexed) plus imatinib vs the measured effects of imatinib/chemotherapeutic combinations were evaluated by the Student’s t test.
In all statistical evaluations the significance threshold is specified in the text.
RESULTS
Expression of PDGFRβ, c-Kit and c-Fms by MMe cells
We evaluated the expression of PDGFRβ, c-Kit (tyrosine kinase receptor for stem cell factor) and c-Fms (M-CSF receptor) by immunoprecipitation and immunoblotting analysis on a panel of eight MMe cell lines. Five of the eight cell lines were positive for PDGFRβ (see fig 1 in supplementary data file available online at http://thorax.bmj.com/supplemental). Of the PDGF receptors, only PDGFRβ (but not PDGFRα) was expressed in MMe cells examined. We selected three MMe cell lines for their different representative expression pattern. PDGFRβ was expressed at a higher level in MMP and REN cells than in ISTMES2 cells, while untransformed HMC did not express the PDGFRβ receptor. The expression of c-Kit and c-Fms occurred at higher levels in MMP cells and was reduced in REN and ISTMES2 cells. HMC only displayed a very low level of c-Fms expression (fig 1A).
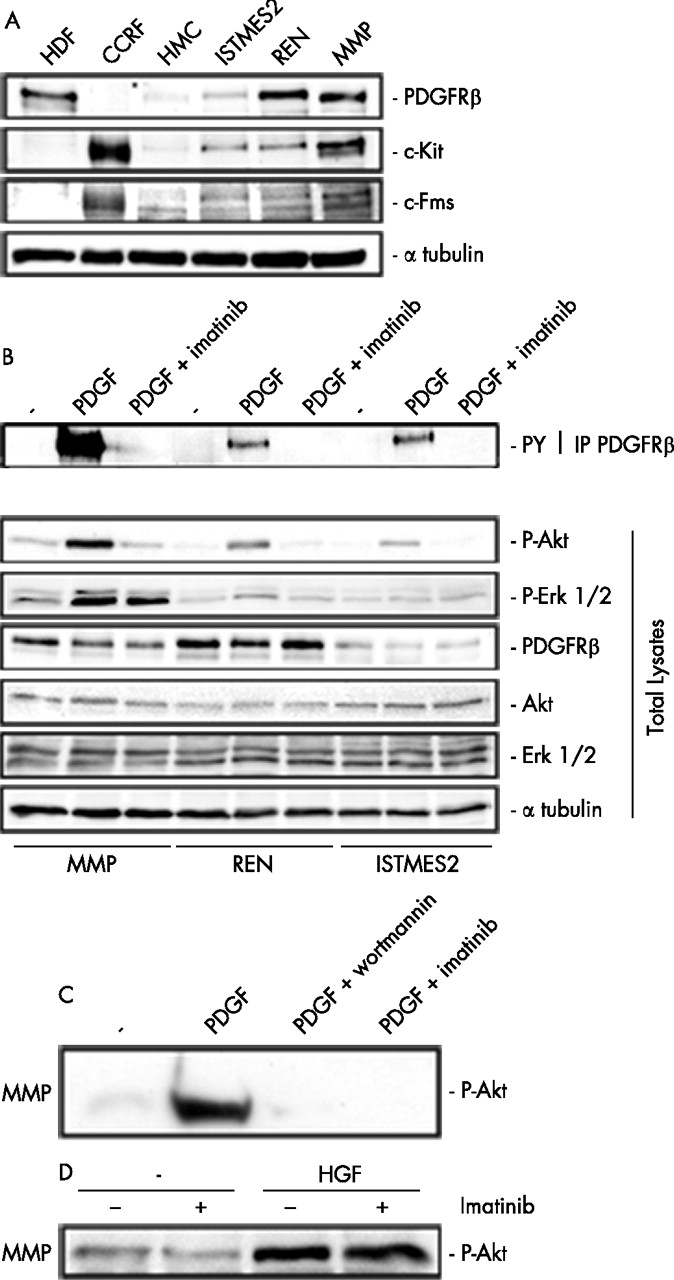
Expression of platelet derived growth factor receptor β (PDGFRβ) in malignant mesothelioma (MMe) cells. (A) Immunoblotting with PDGFRβ, c-Kit and c-Fms antibodies on human mesothelial cells (HMC) and three representative MMe cell lines. Controls: human dermal fibroblasts (HDF) expressing PDGFRβ and CCRF, CCRF-HSB-2, human leukaemic lymphoblast cells expressing c-Kit and c-Fms. (B) Immunoprecipitation with PDGFRβ antibodies followed by immunoblotting with phosphotyrosine antibodies (upper panel); immunoblotting with the indicated antibodies on whole lysates (lower panel). In both panels MMP, REN and ISTMES2 cells were in low serum (−) or stimulated with PDGF in the presence or absence of 10 μM imatinib or (C) 100 nM wortmannin. (D) Immunoblotting with P-Akt (P-Ser 473) antibodies of MMP cells stimulated by 50 ng/ml hepatocyte growth factor (HGF) in the presence or absence of 10 μM imatinib.
Effect of imatinib-mediated PDGFRβ inhibition on Akt
MMe cells positive for PDGFRβ were also tested by immunoprecipitation with PDGFRβ antibodies followed by immunoblotting with phosphotyrosine antibodies after growing cells in low serum conditions. MMe cells displayed negligible levels of tyrosine phosphorylation whereas the addition of recombinant PDGF B increased the receptor phosphorylation of all cells (fig 1B, upper panel). Neither c-Kit nor c-Fms phosphorylation was detectable in the MMe cells (data not shown).
We then determined whether treatment with imatinib could interfere with signalling pathways elicited by this receptor. In low serum conditions, only MMP cells displayed autonomous Akt activity (determined as Ser473 phosphorylation) whereas, on stimulation with PDGF, tyrosine phosphorylation of PDGFRβ and Akt phosphorylation were increased but were markedly inhibited by 10 μM imatinib in all MMe cells examined. Basal Erk1/2 activity was slightly enhanced after PDGF in MMP and, to a lesser extent, in REN cells, while both activities were barely affected by treatment with imatinib 10 μM (fig 1B, lower panel).
Conversely, Akt inhibition was complete and comparable to that obtained by treatment with the phosphatidylinositol-3 kinase (PI3K) inhibitor wortmannin at a concentration of 100 nM (fig 1C). Interestingly, Akt activity in MMP cells, expressing also HGFR/Met,5 was increased by the addition of recombinant HGF (100 ng/ml) but was not affected by imatinib (fig 1D). This indicates a selective blockade of PDGFRβ-dependent Akt signalling by imatinib.
Effect of imatinib on viability of MMe cells expressing PDGFRβ
In view of the crucial role played by Akt in determining survival of HMC and MMe cells,21 we postulated that imatinib could negatively affect the viability of PDGFRβ-positive MMe cells. After incubation for 48 h with up to 100 μM imatinib, cell viability tested by the MTT assay was markedly decreased with a LC50 of 1.84×10−5 M, 1.89×10−5 M and 2.05×10−5 M for MMP, REN and ISTMES2 cells, respectively. Gemcitabine and pemetrexed have already been shown to be particularly effective in combination with cisplatin for MMe chemotherapy.29 We therefore tested the cytotoxic effect induced by these two agents in the presence of different concentrations of imatinib. As expected, gemcitabine and pemetrexed killed MMe cells in a dose-dependent manner. The presence of imatinib modified the profile of the dose-response curves with a shift towards lower LC50 values and a decrease in the fraction of drug-resistant cells (fig 2A).

{kind=link}
{kind=link}
Synergy of imatinib with gemcitabine and pemetrexed. (A) Dose-effect curves of cell viability for gemcitabine (left) and pemetrexed (right) in the presence of different concentrations of imatinib. For the imatinib/gemcitabine combination: •, 1×10−7 M; ▴, 2.5×10−6 M; ▾, 1×10−4 M. For the imatinib/pemetrexed combination: •, 3×10−7 M; ▴, 6×10−7 M; ▾, 1.5×10−6 M. Representative data from three experiments conducted with eight determinations for each point. Points represent mean (±SE) percentage. (B–D) 50% isobologram plot for imatinib in combination with gemcitabine (left) and pemetrexed (right) on MMP, REN and ISTMES2 cells. Points are mean (±SE) 50% lethal concentration (LC50) calculated by regression analysis.
We did not observe any evidence of PDGFRβ phosphorylation/activation by either gemcitabine or pemetrexed (see fig 2 in supplementary data file available online at http://thorax.bmj.com/supplemental), as recently reported for epidermal growth factor receptor.33
Synergy of imatinib with gemcitabine and pemetrexed in inducing MMe cell death
Activation of tyrosine kinase receptors by ligands induces PI3K and Akt activities, exerting several biological effects including increased cell survival with relevant effects on human carcinogenesis.34 We have recently shown that Akt plays a major survival role for MMe cells.21 Therefore, based on the clear-cut toxic effect induced by imatinib on MMe cells mediated by the inhibition of the PI3K/Akt pathway, we hypothesised that this inhibitor may also reinforce cytotoxicity generated by other cytotoxic agents.
Combined treatments of imatinib with other chemotherapeutic drugs were therefore analysed by the isobologram plot method.31 Interestingly, only imatinib/gemcitabine and imatinib/pemetrexed combinations showed a significant synergism in reducing MMP (p⩽0.001) and REN (ranging from p⩽0.01 to p⩽0.001) cell viability compared with the effects observed with single agents alone. This was revealed by inserting all LC50 values on a concave upward curve below the isoeffective plot (fig 2B). In REN cells the synergistic effect is still appreciable, although to a lower extent, while in ISTMES2 cells the effect of imatinib in combination with other chemotherapeutic agents was significantly antagonistic (ranging from p⩽0.05 to p⩽0.001).
The effectiveness of these combined treatments was confirmed when cell death was investigated by TUNEL analysis. The combination of imatinib with gemcitabine or pemetrexed induced a significant increase in apoptosis (p⩽0.001) compared with the theoretical additive effect of each chemotherapeutic agent with imatinib (table 1). No synergistic effect was observed with any of the other chemotherapeutic drugs (results not shown). Interestingly, the concentrations of the single agents used in the combined treatment were much lower than those obtained at therapeutic dosages.
TUNEL analysis of apoptosis induced in malignant mesothelioma (MMe) cells by single drugs or by drug combinations
DISCUSSION
We describe here some preclinical results which provide a rationale for a novel combined approach to MMe treatment via inhibition of PDGFRβ signalling. Our findings on cultured cells are in accordance with previous evidence of PDGFRβ expression in MMe cells and a lack of expression in normal HMC.10 With regard to the relevance of PDGFRβ expression in vivo, the percentage of positive specimens reported is in the range of 30–45% in the different studies reported.24,25 These data therefore offer a rationale for testing the tyrosine kinase inhibitor imatinib on PDGFRβ activity in MMe cells.
Autocrine or paracrine mechanisms may explain the activation of PDGFRβ in vivo. An autocrine loop has been described as an activating mechanism leading to tyrosine kinase receptor activity in MMe cells,5 and the stromal microenvironment has been shown to be a fundamental source of activating ligands for PDGFR in human tumours.35 Tyrosine phosphorylation of this receptor in MMP and REN cells is inhibited by imatinib, leading to cytotoxic effects and addressing the role of downstream PI3K/Akt survival signalling.
We and others have shown that Akt activation in MMe cells is a crucial signalling pathway which contributes to the MMe malignant phenotype.21,36 Even though this is also dependent on the activity of several other tyrosine kinase receptors, our findings show that specific interference with the PDGFRβ-dependent pathway causes an increase in cell chemosensitivity.
Preclinical studies on several human solid tumours have confirmed the efficacy of imatinib as a cytotoxic agent.37–40 In chronic myeloid leukaemia and gastrointestinal stromal tumour the carcinogenic role of the fusion protein BCR-ABL39 and activating mutations of c-Kit,41 respectively, predict a clinical response to imatinib. Conversely, in MMe, two recent negative reports gave clear evidence that imatinib monotherapy is ineffective.25,26 On the other hand, combined treatment with imatinib and different chemotherapeutic agents has been shown to be effective in mice.27,28
Our results clearly indicate that PDGFRβ expression in MMe cells is essential for sensitivity to imatinib and for the synergy observed between imatinib and gemcitabine or pemetrexed. However, when all three receptors sensitive to imatinib and upstream of the PI3K/Akt pathway are co-expressed in the same cell type, as in MMP, the synergistic effect is higher than in REN cells where only two of them are expressed (PDGFRβ and c-Kit).
Gemcitabine and pemetrexed are known to be active on MMe cells42 and their combination with imatinib has intriguing implications. In particular, the synergism shown in this study indicates that very low doses of chemotherapeutic agents should be sufficient to exert a therapeutic effect.
Given our previous findings,21 the mechanism underlying the observed in vitro synergy is probably imatinib-dependent PDGFRβ inhibition which, in turn, leads to Akt inactivation resulting in sensitisation of MMe cells to low chemotherapeutic concentrations. However, it is conceivable that other biological effects could play a role in humans. Reduction of the intratumoral interstitial fluid pressure and increased uptake of chemotherapeutics by imatinib have been shown in vivo.27 In addition, imatinib has been shown to interfere with VEGF expression and associated neoangiogenesis.43
Although some progress has been made in the treatment of MMe, the results are still unsatisfactory and MMe remains an ideal field in which to test new therapeutic approaches.42 Our work on the synergy between imatinib and chemotherapeutic drugs active in MMe provides a strong rationale for a new approach to the treatment of this disease and will be evaluated further in early phase clinical trials.
Acknowledgments
The authors thank Dr Patrizia Morbini, Institute of Pathology, San Matteo University Hospital, Pavia, Italy and also Dr M Rinaldi for advice on statistical analysis.
REFERENCES
Supplementary materials
Files in this Data Supplement:
Footnotes
-
Published Online First 22 February 2007
-
GG and LM contributed equally to the study.
-
This work was supported by research grants to GG from AIRC (Associazione Italiana per la Ricerca sul Cancro), MARF (Mesothelioma Applied Research Foundation) and the Buzzi Foundation (Casale Monferrato, Italy). This work is part of G.I.Me. (Gruppo Italiano per lo Studio e la Terapia del Mesotelioma) network program.
-
Competing interests: None declared.