Article Text

Abstract
Background: The angiotensin system has a role in the pathogenesis of pulmonary fibrosis. This study examines the antifibrotic effect of losartan, an angiotensin II type 1 receptor antagonist, in bleomycin induced lung fibrosis and its possible implication in the regulation of prostaglandin E2 (PGE2) synthesis and cyclooxygenase-2 (COX-2) expression.
Methods: Rats were given a single intratracheal instillation of bleomycin (2.5 U/kg). Losartan (50 mg/kg/day) was administrated orally starting one day before induction of lung fibrosis and continuing to the conclusion of each experiment.
Results: Losartan reduced the inflammation induced by bleomycin, as indicated by lower myeloperoxidase activity and protein content in the bronchoalveolar lavage fluid. Collagen deposition induced by bleomycin was inhibited by losartan, as shown by a reduction in the hydroxyproline content and the amelioration of morphological changes. PGE2 levels were lower in fibrotic lungs than in normal lungs. Losartan significantly increased PGE2 levels at both 3 and 15 days. A reduction in COX-2 expression by bleomycin was seen at 3 days which was relieved by losartan.
Conclusions: The antifibrotic effect of losartan appears to be mediated by its ability to stimulate the production of PGE2. Losartan, which is already widely used clinically, could be assessed as a new treatment in lung fibrosis.
- ACE, angiotensin converting enzyme
- ANGII, angiotensin II
- AT1, angiotensin type 1 receptor
- COX-2, cyclooxygenase-2
- HP, hydroxyproline
- IPF, idiopathic pulmonary fibrosis
- MPO, myeloperoxidase
- PGE2, prostaglandin E2
- TGF-β, transforming growth factor β
- pulmonary fibrosis
- cyclo-oxygenase
- COX-2
- angiotensin II
- losartan
- prostaglandin E2
Statistics from Altmetric.com
- ACE, angiotensin converting enzyme
- ANGII, angiotensin II
- AT1, angiotensin type 1 receptor
- COX-2, cyclooxygenase-2
- HP, hydroxyproline
- IPF, idiopathic pulmonary fibrosis
- MPO, myeloperoxidase
- PGE2, prostaglandin E2
- TGF-β, transforming growth factor β
Idiopathic pulmonary fibrosis (IPF) is a specific form of chronic interstitial lung disease which is associated with the histological appearance of usual interstitial pneumonia. The poor prognosis of IPF patients, with a mean survival period of 2–4 years, and the inefficacy of current treatment based on corticosteroids and immunosuppressive drugs, underlines the need for new therapeutic strategies.1 IPF is characterised by the loss of lung architecture through increased epithelial cell apoptosis and abnormal wound repair, leading to the formation of fibroblast-myofibroblast foci and extracellular matrix deposition.2–4 This pathological process is related to the interactions of several cytokines, chemical mediators, and growth factors derived from epithelial and mesenchymal cells.5,6 Experimental lung fibrosis induced by bleomycin is a well studied model of fibrogenesis supported by ample literature. This model of pulmonary fibrosis resembles that seen in humans and has been used to assess the effects of potential therapeutic agents.
Angiotensin II (ANGII) is produced by proteolytic cleavage of its precursor angiotensin I by angiotensin converting enzyme (ACE). Experimental evidence suggests that ANGII regulates the fibrotic response to tissue injury. It has been reported that ANGII plays an important role in cardiac, renal, hepatic, and pancreatic fibrogenesis.7,8,9,10 It has also been shown that ANGII is a crucial mediator in the pathogenesis of pulmonary fibrosis. ANGII induces proliferation of human lung fibroblasts and production of lung procollagen via activation of angiotensin type 1 receptor (AT1).11–13
It is known that losartan, a selective AT1 receptor antagonist, inhibits the proliferation of human lung fibrotic fibroblasts induced by ANGII in vitro.12,13 Moreover, losartan inhibits the deposition of lung collagen in the rat model of bleomycin induced pulmonary fibrosis.11
The mechanisms involved in the pulmonary antifibrotic effect of losartan have been widely investigated but are still not fully understood. One potential mechanism is the regulation of other inflammation mediators and growth factors. There is evidence that the antifibrotic effect of losartan is mediated through AT1 receptors and involves at least the downregulation of transforming growth factor β (TGF-β), a profibrotic mediator.11
It has been reported that prostaglandin E2 (PGE2) is a potent inhibitor of fibroblast proliferation, collagen synthesis, and fibroblast to myofibroblast differentiation.4 Bronchoalveolar lavage (BAL) fluid from patients with IPF has been found to contain 50% less PGE2 than normal individuals.14 Furthermore, alveolar macrophages and fibroblasts have a reduced capacity to produce PGE2 in vitro.15,16 The failure to synthesise PGE2 has been shown to be associated with a decreased capacity to upregulate cyclo-oxygenase 2 (COX-2).16–18
The main objective of this study was to investigate the antifibrotic effect of losartan and its possible implication in the regulation of PGE2 synthesis and COX-2 expression in a rat model of bleomycin induced pulmonary fibrosis. Some of the results of this study have previously been reported in the form of abstracts.19,20
METHODS
Animals
Adult male pathogen free Sprague-Dawley rats weighing 225–250 g at the beginning of the studies were obtained from Criffa (Iffa Credo, France). The animals were maintained in a controlled environment and fed on rodent chow (A04; Panlab, Barcelona, Spain) and tap water ad libitum.
Chemicals
Bleomycin sulphate was obtained from Almirall-Prodesfarma (Barcelona, Spain), halothane (Fluothane) from Zeneca Farma (Pontevedra, Spain), and sodium pentobarbital from Normon (Madrid, Spain). Losartan was provided by Merk and Co (West Point, PA, USA).
Experimental model
Animals were anaesthetised under halothane and a single dose of 2.5 U/kg bleomycin dissolved in sterile saline (0.9% NaCl) was instilled intratracheally via the transoral route by a small glass device developed especially for it.21 Control animals received the same volume of intratracheal saline solution. The dose of bleomycin was selected from previous experiments to cause no mortality but consistent biochemical and histological damage.21,22 The duration of each experiment was 3 or 15 days post-instillation. The animals were sacrificed at the end of each experiment by a lethal injection of sodium pentobarbital (100 mg/kg ip) followed by exsanguination from the abdominal aorta. Lung tissues were weighed and processed separately for bronchoalveolar lavage, biochemical, and histological studies as described below.
Experimental groups
Animals were randomly distributed into four groups in each experiment: (1) sterile saline solution (vehicle for bleomycin) + water (vehicle for losartan) (n = 4); (2) sterile saline solution + losartan (n = 4); (3) bleomycin + water (n = 8); (4) bleomycin + losartan (n = 8).
Losartan (50 mg/kg/day) or water was administered orally (at 09.00 hours) starting one day before the instillation of bleomycin and continuing up to the end of the experiment. The treatment was dissolved in a final volume of 1–2 ml distilled water. The experimental groups were repeated twice at 3 days and at 15 days. Oral administration was selected as this is usual in the clinical setting, and the dose level and schedule were based on previous studies.
Biochemical studies
Neutrophilic infiltration was assessed by measuring myeloperoxidase (MPO) activity.23 MPO was measured photometrically with 3,3′,5,5′-tetramethylbenzidine as a substrate. Lung samples were macerated with 0.5% hexadecyltrimethylammonium bromide in 50 mM phosphate buffer at pH 6.0. Lung homogenates were disrupted for 30 seconds using a Labsonic sonicator (B Braun) at 20% power and submitted to three cycles of snap freezing in dry ice and thawing before a final 30 second sonication. Samples were incubated at 60°C for 2 hours and then spun down at 4000g for 12 minutes. The supernatants were collected for MPO assay. Enzyme activity was assessed photometrically at 630 nm. The assay mixture consisted of 20 µl supernatant, 10 µl tetramethylbenzidine (final concentration 1.6 mM) dissolved in DMSO, and 70 µl H2O2 (final concentration 3.0 mM) diluted in 80 mM phosphate buffer, pH 5.4. The results are expressed as units (U) MPO activity per lung.
Lung hydroxyproline (HP) content was measured as an indicator of collagen deposition by the method outlined by Woessner.24 Lung samples were homogenised and then hydrolysed in 6 N HCl for 18 hoursat 110°C. The hydrolysate was neutralised with 2.5 M NaOH. Aliquots (2 ml) were analysed for HP content after the addition of chloramine T (1 ml), perchloric acid (1 ml), and dimethylaminobenzaldehyde (1 ml). Samples were read for absorbance at 550 nm in a spectrophotometer. The results are expressed as μg HP per lung.
Bronchoalveolar lavage fluid
BAL fluid was obtained by cannulating the trachea and lavaging four times with 10 ml NaCl 0.9%. The BAL fluid was centrifuged (300g, 10 minutes, 4°C) and the supernatant was used for biochemical studies. Total protein concentration was measured by a standard dye technique (BioRad; Munich, Germany). The levels of PGE2 were measured by a competitive enzyme immunoassay kit (Cayman Chemical; Ann Arbor, MI, USA) following the manufacturer’s protocol.
RNA preparation and RT-PCR of COX-2
Total RNA was prepared with Trizol reagent following the manufacturer’s instructions (GibcoBRL, Life Technologies) and RNA concentrations were calculated from A260 determinations. RNA integrity and loading amounts were assessed by examining 18S and 28S ribosomal RNA banding of samples electrophoresed in 1% agarose gel under non-denaturing conditions and stained with ethidium bromide. Analysis of COX-2 and β-actin mRNA expression was done using a semi-quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) method. One µg total RNA was used and the sequences amplified by the Life Technologies One Step RT-PCR System according to the manufacturer’s protocol. The forward primer for COX-2 was 5′-GCT-GTA-CAA-GCA-GTG-GCA-AA-3′ and the reverse primer was 5′-ATG-GTG-GCT-GTC-TTG-GTA-GG-3′. For β-actin the forward primer was 5′-TCA-TGA-AGT-GTG-ACG-TTG-ACA-TCC-GT-3′ and the reverse primer was 5′-CCT-AGA-AGC-ATT-TGC-GGT-GCA-CGA-TG-3′. Sequences were resolved by electrophoresis in denaturing 1.8% agarose gel and staining with ethidium bromide.
Lung histological studies
Histological evaluation was carried out on lungs that were not lavaged. The lungs were first perfused through their main bronchus with a fixative solution (10% neutral buffered formalin) at a pressure of 25 cm H2O, immersed in the fixative for 12–24 hours, and blocks were taken. Tissue paraffined blocks were cut into 4 μm thick sections and stained with haematoxylin-eosin and Masson’s trichrome to identify inflammatory cells, connective tissue, and collagen deposition. The histological changes were evaluated by a descriptive method.
Statistical analysis
Data are expressed as mean (SE) with 95% confidence intervals (CI) of n experiments. Statistical analysis was carried out by analysis of variance (ANOVA) followed, when differences were significant, by appropriate post hoc tests including the Newman-Keuls tests. Differences between groups were tested using the paired Student’s t test (GraphPad Software Inc, San Diego, CA, USA). Differences were considered statistically significant when p was <0.05.
RESULTS
Effect of losartan on bleomycin induced lung inflammation and collagen deposition
The mean (SE) lung/body weight ratio was significantly increased in fibrotic rats induced by bleomycin both at 3 days (control group: 0.0045 (SE 0.0001) (95% CI 0.0046 to 0.0047); bleomycin group: 0.0077 (SE 0.0003) (95% CI 0.0071 to 0.0083)) and at 15 days (control group: 0.004 (SE 0.0002) (95% CI 0.0037 to 0.0043); bleomycin group: 0.0084 (SE 0.0012) (95% CI 0.0071 to 0.0098)). Losartan treatment did not have a significant effect on this ratio (fig 1A and B).

Relationship between lung weight and body weight at (A) 3 days and (B) 15 days. There was a significant increase in the lung/body weight relationship after bleomycin (BLM) administration at 3 days (*p = 0.001 versus control group) and at 15 days (*p<0.001 versus control group). Losartan (LOS) had no effect on this relationship (3 days: p = 0.84; 15 days: p = 0.88). Statistical analysis was performed by one way ANOVA followed by Student’s t test.
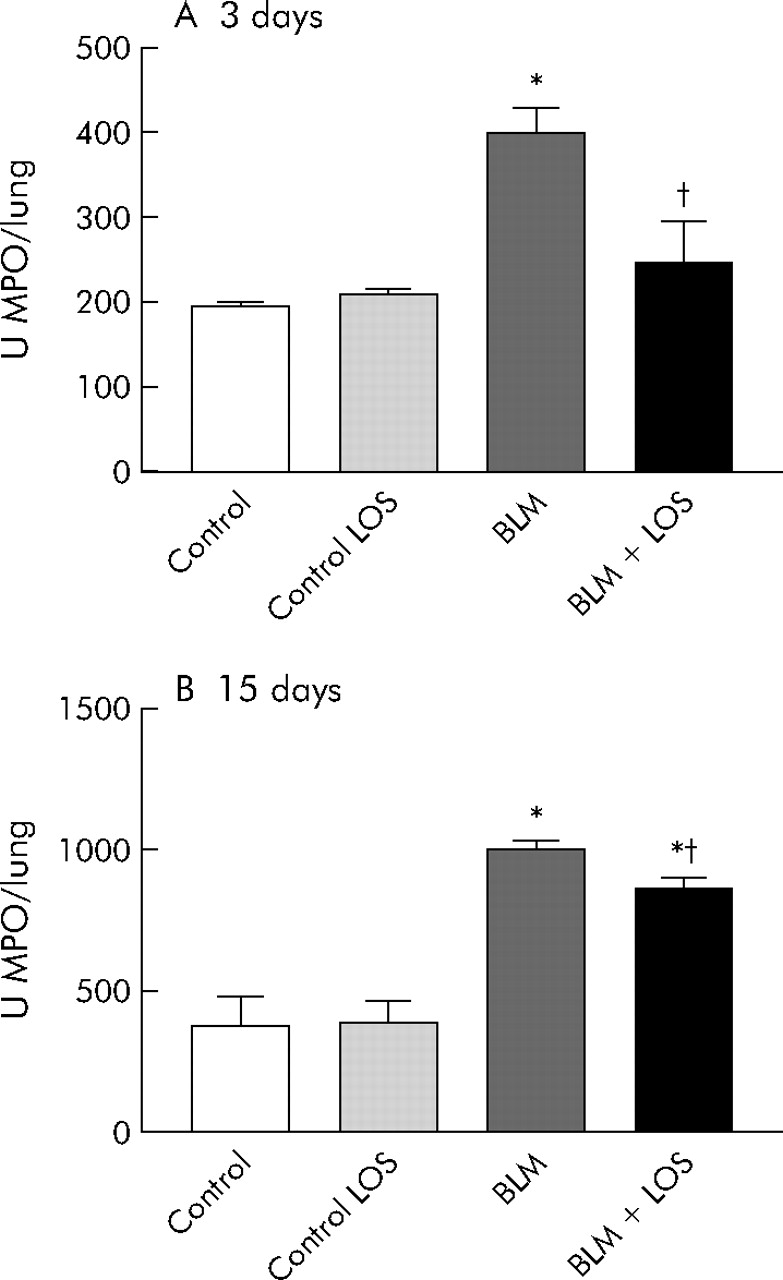
An increase in MPO activity was observed in bleomycin exposed rats compared with the control groups at 3 and 15 days (fig 2A and B) after bleomycin instillation (3 days: control group 146.8 (5.2) U/lung (95% CI 138.4 to 155.1), bleomycin group 399 (28.7) U/lung (95% CI 350.3 to 447.7); 15 days: control group 279.5 (103.9) U/lung (95% CI 177.6 to 381.4), bleomycin group 1000 (16.4) U/lung (95% CI 968 to 1032)). This increase was significantly reduced by losartan (3 days: bleomycin + losartan group 247.1 (28.6) U/lung (95% CI 198.5 to 295.7), 15 days: bleomycin + losartan group 869.4 (24.9) U/lung (95% CI 832.1 to 906.7)).
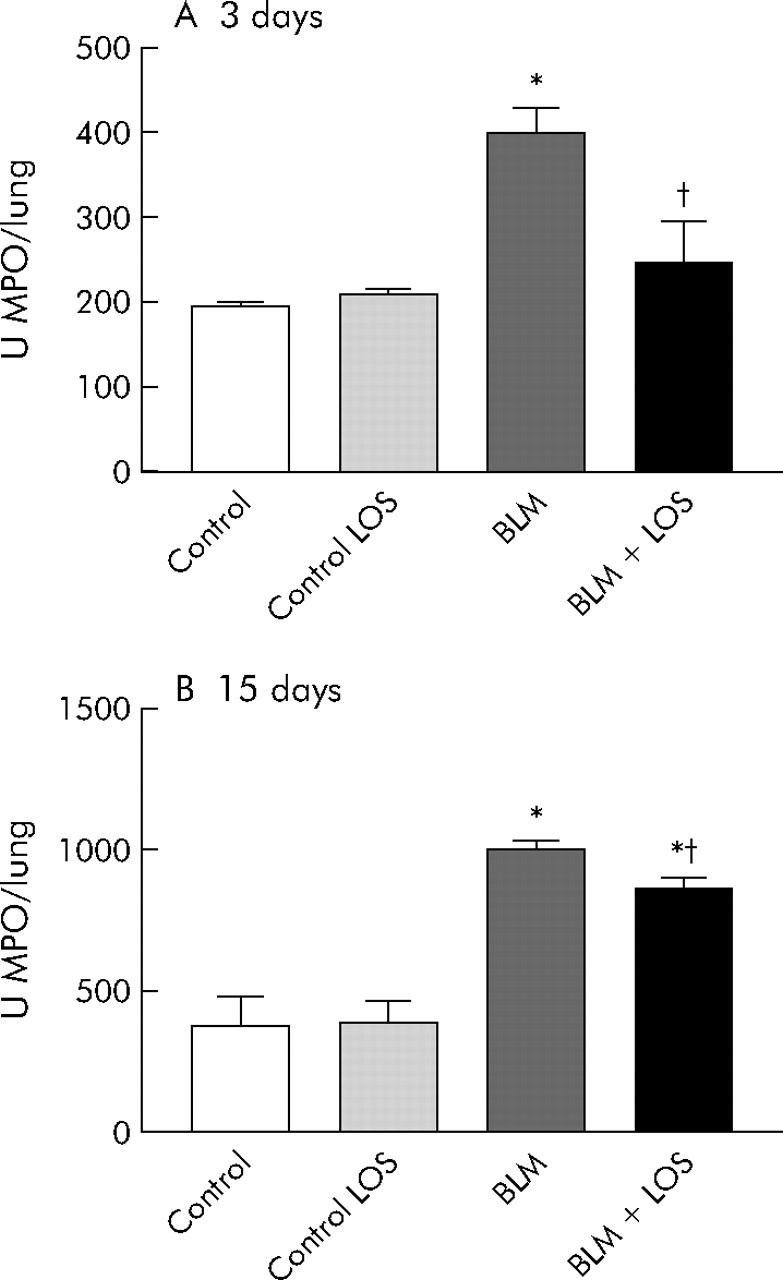
Effect of losartan (LOS) on lung inflammation induced by bleomycin (BLM) assessed by myeloperoxidase (MPO) activity (U/lung) in lung tissue at (A) 3 days and (B) 15 days. Bleomycin induced a rise in MPO activity at both 3 and 15 days, and this increase was inhibited by losartan (50 mg/kg). Statistical analysis was performed by one way ANOVA followed by Student’s t test. *p<0.001 versus control group; †p = 0.004 versus BLM group at 3 days; †p = 0.005 versus BLM group at 15 days.
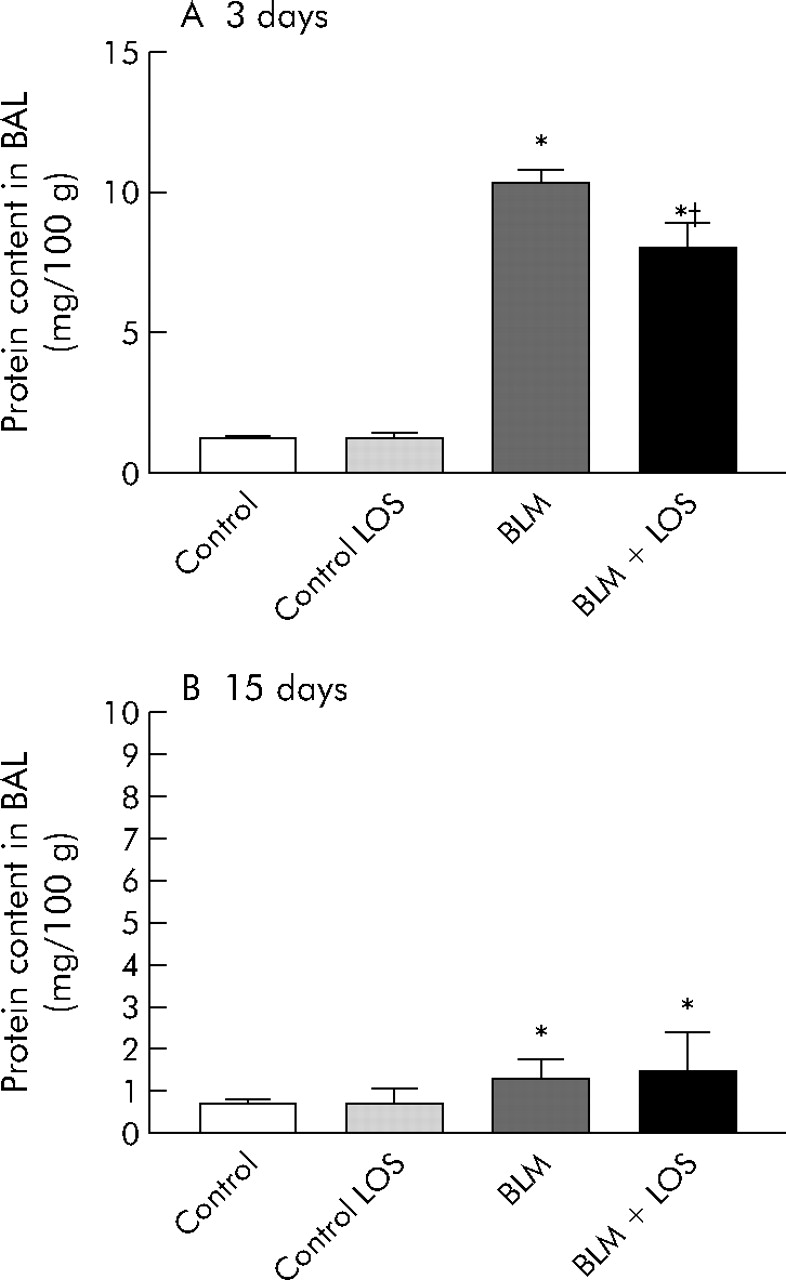
Total recovery of BAL fluid typically exceeded 80%, and the percentages of fluid recovered did not differ significantly between experimental groups. Protein in BAL fluid was significantly increased after 3 days of bleomycin instillation, and this effect was inhibited by losartan treatment (control group: 1.2 (0.1) mg/100 g (95% CI 1 to 1.4); bleomycin group: 10.3 (0.5) mg/100 g (95% CI 9.4 to 11.2); bleomycin + losartan group: 8 (0.9) mg/100 g (95% CI 6.5 to 9.5), fig 3A). Fifteen days after bleomycin instillation the difference between the control group and bleomycin group was significant, but there were no differences between the bleomycin group and the losartan treatment group (control group: 0.7 (0.1) mg/100 g (95% CI 0.6 to 0.8); bleomycin group: 1.3 (0.1) mg/100 g (95% CI 1.1 to 1.5), fig 3B).
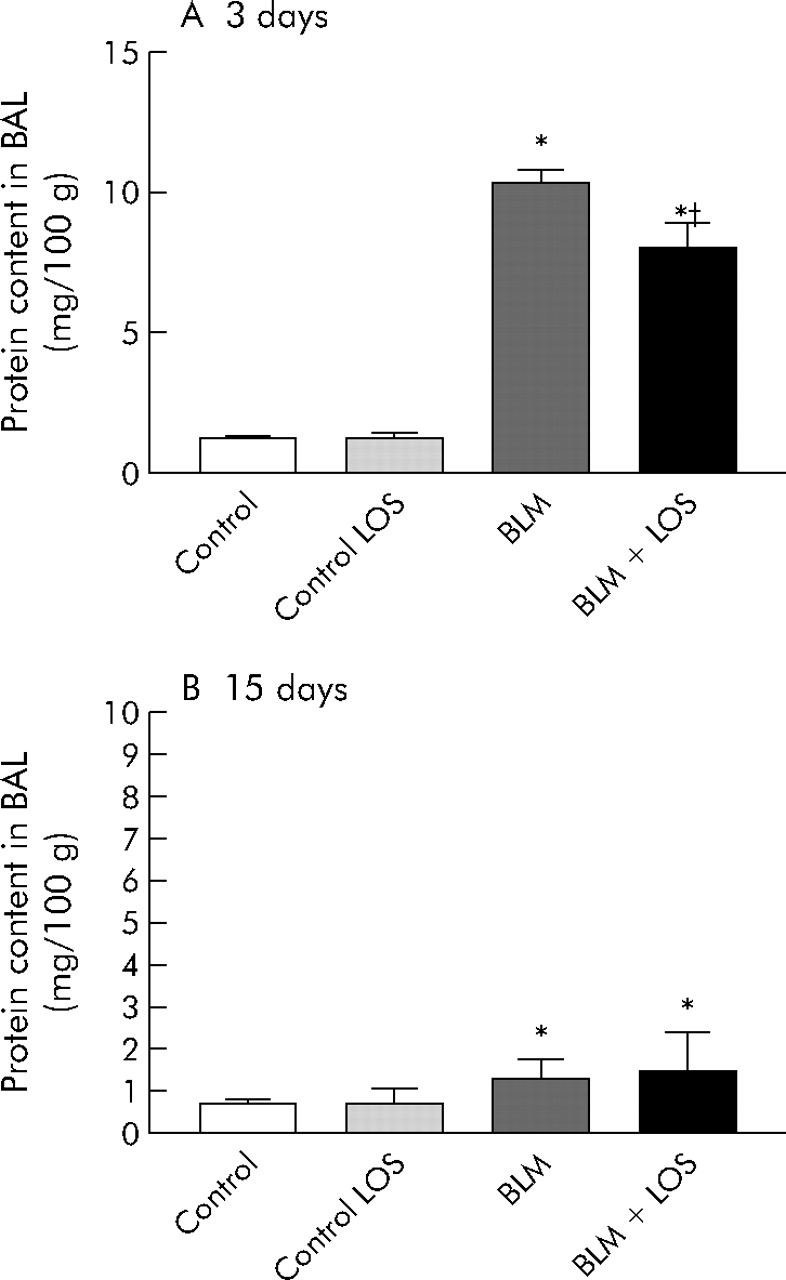
Effect of losartan (LOS) on lung inflammation induced by bleomycin (BLM) measured by protein content in BAL fluid (mg/100 g) at (A) 3 days and (B) 15 days. Protein in BAL fluid increased after BLM instillation on days 3 and 15 (*p<0.001 versus control group at 3 days; *p = 0.04 versus control group at 15 days). At 3 days losartan reduced the total protein production initially induced by BLM (†p = 0.04 versus BLM group). No differences were found between the BLM group and BLM + LOS group at 15 days (p = 0.11, fig 3B). Statistical analysis was performed by one way ANOVA followed by Student’s t test.
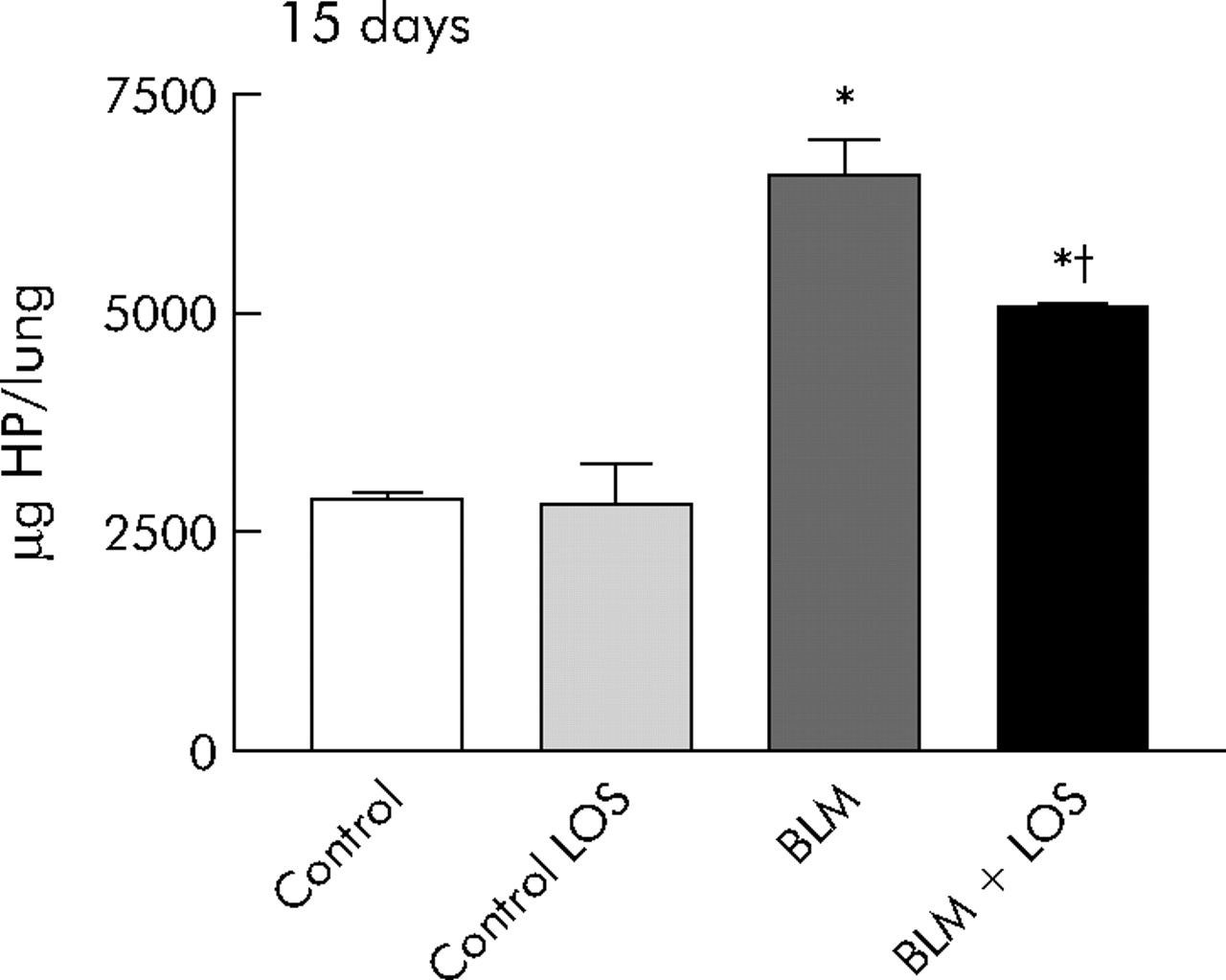
Lung HP content, a marker of collagen deposition, was increased at 15 days after exposure to bleomycin. This increase was significantly blocked by losartan, although levels remained higher than those found in animals not exposed to bleomycin (control group: 2875 (71) μg/lung (95% CI 2794.5 to 2955); bleomycin group: 6564 (414) μg/lung (95% CI 5838.4 to 7290); bleomycin + losartan group: 5081 (43) μg/lung (95% CI 5021.6 to 5140.6), fig 4).

Antifibrotic effect of losartan (LOS) on bleomycin (BLM) induced lung fibrosis quantitatively measured using hydroxyproline (HP) content (μg/lung) at 15 days. The increased amount of HP content after BLM induction was inhibited by LOS. Statistical analysis was performed using one way ANOVA followed by Student’s t test: *p = 0.001 versus control group; †p<0.001 versus BLM group.
Effect of losartan on PGE2 and COX-2 mRNA levels
The concentration of PGE2 in BAL fluid (pg/lung) was significantly inhibited by bleomycin instillation at 3 days. This effect was abrogated by losartan treatment which increased the level of PGE2 (control group: 33849.1 (2065.6) pg/lung (95% CI 30985.9 to 36712.2); bleomycin group: 24673.7 (1516.7) pg/lung (95% CI 22015 to 27332.5); bleomycin + losartan group: 35076.3 (3986.6) pg/lung (95% CI 28309.6 to 41843.1), fig 5A). There were no significant differences in the amount of PGE2 between control and BLM groups 15 days after fibrotic induction. Losartan treatment significantly increased the PGE2 concentration at 15 days (control group: 63520.3 (9189.5) pg/lung (95% CI 47922.1 to 79118.4); bleomycin group: 64499.6 (2807) pg/lung (95% CI 59735 to 69264.1); bleomycin + losartan group: 81419.1 (5542.5) pg/lung (95% CI 71361.8 to 91476.4), fig 5B).

Effect of losartan (LOS) on synthesis of prostaglandin E2 (PGE2, pg/lung) at (A) 3 days and (B) 15 days after bleomycin (BLM) instillation. PGE2 synthesis was reduced at 3 days and this reduction was prevented by losartan (*p = 0.01 versus control group; †p = 0.01 versus BLM group). At 15 days rats instilled with saline solution or BLM showed no differences in PGE2 synthesis (p = 0.9), but LOS treatment upregulated PGE2 production (†p = 0.004 versus BLM group). Statistical analysis was performed using one way ANOVA followed by Student’s t test.
The expression of COX-2 mRNA was generally lower 3 days after bleomycin instillation. Fibrotic rats treated with losartan expressed a significantly increased amount of COX-2 mRNA at 3 days (control group: 0.44 (0.05) (95% CI 0.35 to 0.52); bleomycin group: 0.38 (0.003) (95% CI 0.37 to 0.38); bleomycin + losartan group: 0.59 (0.03) (95% CI 0.54 to 0.64), fig 6A). There were no changes in COX-2 mRNA expression at 15 days after bleomycin instillation and no significant differences were observed in the losartan group (control group: 0.53 (0.01) (95% CI 0.45 to 0.62); bleomycin group: 0.51(0.06) (95% CI 0.37 to 0.64); bleomycin + losartan group: 0.45 (0.08) (95% CI 0.28 to 0.62), fig 6B).

Effect of losartan (LOS) on lung COX-2 mRNA expression at (A) 3 days and (B) 15 days after bleomycin (BLM) instillation. The reduction in COX-2 expression after 3 days of BLM instillation was not significant. An increase in COX-2 expression was observed after 3 days of BLM instillation with LOS treatment (†p = 0.02 versus BLM group). At 15 days there were no differences between the BLM and BLM+LOS groups (p = 0.3). Statistical analysis was done by one way ANOVA followed by Student’s t test.
Histological features
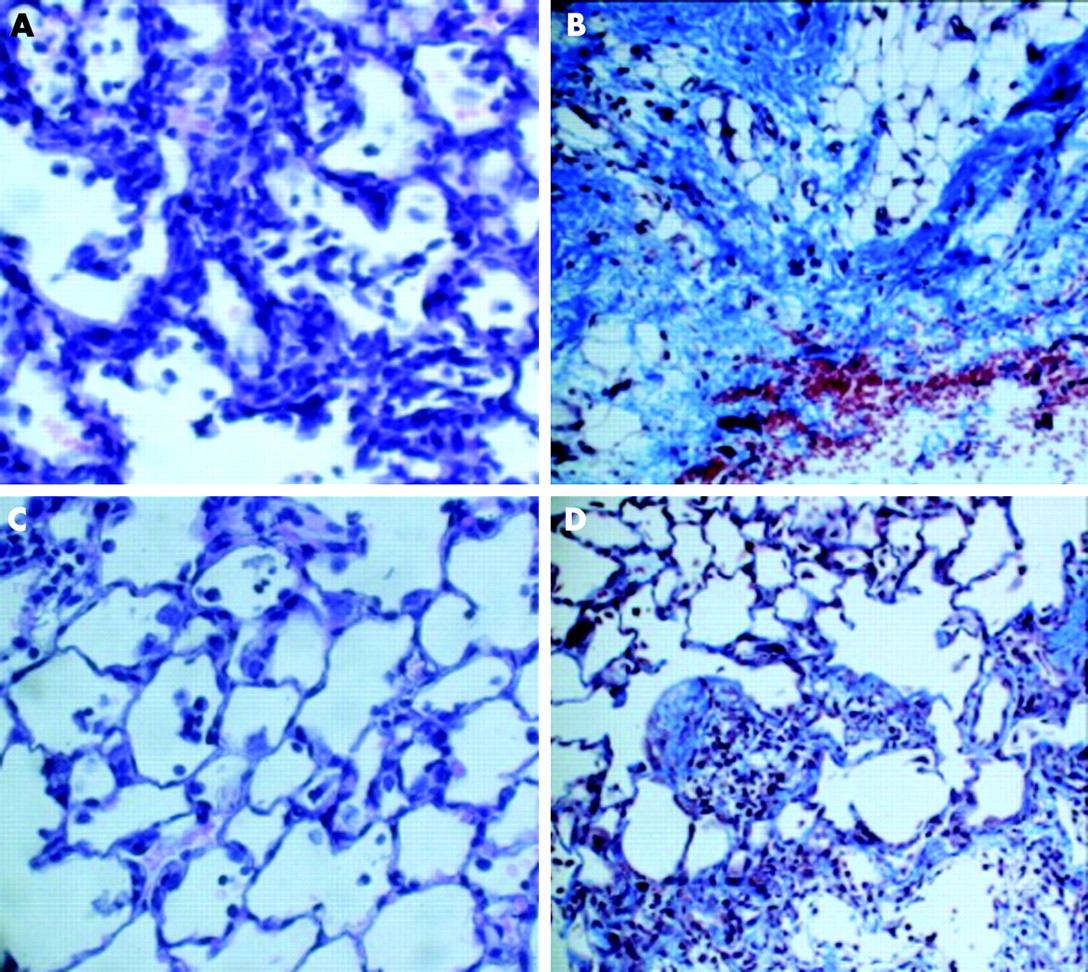
Haematoxylin-eosin and Masson’s trichrome stained lung sections were examined by light microscopy. Lungs from control groups at 3 and 15 days were histologically normal (data not shown). The administration of bleomycin resulted in characteristic histological changes including areas of inflammatory infiltration, thickening of alveolar walls (fig 7A) and an increase in interstitial collagen deposition and fibroblastic appearance (fig 7B). Losartan treatment significantly reduced the changes in lung morphology; there were fewer inflammatory infiltrates (fig 7C), less collagen deposition, and septal widening (fig 7D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histological examination of the anti-inflammatory and antifibrotic effects of losartan on bleomycin instilled lungs. Rats were instilled with bleomycin or saline solution. After 3 or 15 days of treatment with losartan they were sacrificed and the removed lungs stained with haematoxylin-eosin (H-E) or Masson-trichrome stain. (A) Fibrotic lung induced by bleomycin at 3 days and stained with H-E showing extensive interstitial inflammatory infiltration. (B) Fibrotic lung induced by bleomycin at 15 days and stained with Masson showing patchy areas of interstitial collagen deposition. (C) Fibrotic lung treated with losartan at 3 days and stained with H-E showing fewer infiltrates of inflammatory cells. (D) Fibrotic lung treated with losartan at 15 days and stained with Masson: although multifocal parenchymal lesions were also present, there was less septal widening and less collagen deposition. All fields were examined in each group. Original magnification: ×40.
DISCUSSION
We have shown that losartan, a selective AT1 receptor antagonist, ameliorates experimental lung fibrosis induced by bleomycin and increases PGE2 synthesis. Our findings provide further evidence for the antifibrotic effect of losartan and support the potential role of PGE2 as a protective molecule in lung fibrosis.
ANGII has been shown to be an important mediator in the pathogenesis of lung fibrosis and may influence the progression of lung injury via a number of mechanisms.2,5 It has been reported that there is an increase in lung ANGII concentration, which precedes increases in lung collagen, and an upregulation in AT1 receptor expression in lung parenchyma after bleomycin induced lung injury.11,12,25 The presence of a pulmonary renin-angiotensin system within the lung has been suggested several times in the literature. It has been reported that angiotensinogen and AT1 receptors are expressed in lung tissue.26 Moreover, a high concentration of ACE in BAL fluid has been observed in patients with sarcoidosis and IPF.27
The study shows that losartan significantly decreased lung inflammatory infiltration both early (3 days) and late (15 days) after bleomycin exposure. The anti-inflammatory effect of AT1 receptor blockade has been reported in previous studies.25,28,29 Our findings agree with those of Otsuka et al25 who showed that candesartan cilexetil significantly decreases the concentration of neutrophils in BAL fluid in the bleomycin model of pulmonary fibrosis, suggesting that ANGII may play a role in neutrophilic infiltration in the lung. Our study also confirms that losartan is effective in reducing lung fibrosis induced by bleomycin. Controversially, a recent study by Keogh et al30 has reported that losartan cannot attenuate the collagen deposition induced by bleomycin in mice. Differences in the animal model, the dose of bleomycin and losartan, and the time point of HP measurements may account for discrepancies between our results and those of Keogh et al.30 Nevertheless, a large number of previous reports have shown that AT1 receptor antagonists decrease lung collagen deposition, in accordance with our observations.11,25,31
The mechanisms of the pulmonary antifibrotic effect of AT1 receptor antagonists have been investigated in recent years.11–13,25 It is known that losartan inhibits the proliferation of human lung fibrotic fibroblasts induced by ANGII in vitro, through the activation of AT1 receptor and mediated by TGF-β.12 Renzoni et al have reported that the ANGII receptor type 1 gene is one of the most relevant targets of TGF-β in primary lung fibroblasts from patients with IPF.32 In the bleomycin model of pulmonary fibrosis, administration of losartan or candesartan cilexetil attenuates TGF-β expression and lung collagen deposition.11,25 Similarly, other observations suggest that the antifibrotic effect of losartan could involve the regulation of TGF-β expression, providing further insights into the links between profibrotic mediators in the pathogenesis of pulmonary fibrosis. The present study was designed to investigate whether losartan can also act through antifibrotic mediators.
The most relevant finding in our study was the effect of losartan on PGE2 synthesis and COX-2 expression. Losartan significantly reversed the decrease in PGE2 synthesis induced by bleomycin. A growing body of evidence supports the hypothesis that PGE2 has a crucial role in the modulation of tissue repair and lung fibrosis.33 PGE2 inhibits fibroblast migration and proliferation in response to various mitogens and abrogates TGF-β induced collagen production.16,34,35 In addition, fibrotic fibroblasts exhibit a marked reduction in the ability to upregulate PGE2 synthesis in response to TGF-β, with the consequent loss of the antiproliferative response to TGF-β mediated by PGE2.17 Moreover, lung epithelial cells are a major source of PGE2, and the capacity of these cells to inhibit fibroblast proliferation is related to their ability to produce PGE2.36 It appears that these changes are important in transforming the fibroblast from a relatively passive cell into a crucial component of IPF pathogenesis.2 We hypothesised that the increased amount of ANGII may suppress the induction of PGE2 synthesis, and this may result in an imbalance between profibrotic and antifibrotic mediators. Thus, reduction in the activity of ANGII would enhance the protective effect of PGE2 in the fibrotic process. Our results confirm that AT1 antagonism increases PGE2 and reduces collagen deposition. Taken together, these observations indicate that losartan acts on at least two of the most crucial mediators implicated in lung fibrosis, improving the balance between profibrotic (TGF-β1) and antifibrotic (PGE2) mediators.
The analysis of COX-2 mRNA expression showed that losartan reversed the reduction of COX-2 induced by bleomycin at 3 days, but no significant differences were found at 15 days. These results concur with studies in the kidney in which it was shown that ANGII inhibits COX-2 expression in rat renal cortex and that the administration of either captopril or losartan increases this expression.37 However, the mechanisms involved in the regulation of COX-2 by ANGII in pulmonary fibrosis have not been investigated. The relevance of COX-2 as a protective mediator of pulmonary fibrosis has been demonstrated in vivo and in vitro.17,38,39 COX-2 is the major source of the PGE2 synthesised by alveolar epithelial cells.36 The failure to synthesise PGE2 in fibroblasts and lung tissue from patients with IPF has been shown to be associated with a decreased capacity to upregulate COX-2.16–18 Interestingly, the increased PGE2 level observed in lung fibrosis under losartan treatment did not correlate with a significant increase in COX-2 mRNA expression. This could be a consequence of the fact that AT1 antagonists also have an effect on other enzymes implicated in PGE2 synthesis such as PGE2 synthase, a hypothesis which requires future investigation.
In summary, our study shows that losartan enhances PGE2 synthesis in experimental lung fibrosis and provides new insights into how AT1 receptor antagonists act against lung fibrosis. In addition, these data throw new light on the importance of ANGII as a regulator of the other main molecules implicated in the fibrotic process. This is a new approach to the molecular mechanisms implicated in the lung fibrotic process which could provide new insights for future treatments in lung fibrosis.
Acknowledgments
The authors thank Magda Mauri Cugat for her technical assistance and the animal facility carers at the University of Barcelona for their general support.
REFERENCES
Footnotes
-
Published Online First 6 April 2006
-
This study was supported in part by grants from Hospital Clínic de Barcelona (Premi Fi de Residencia, 2003), SEPAR 2003, FIS 02/0186, FUCAP 2004, Red Respira (Fondo de Investigaciones Sanitarias, V-2003-REDC11D-0), and I+D SAF2003-04450.
-
Competing interests: none.
-
The study was approved by the institutional ethics committee and complied with European Community regulation (Directive 86/609/EEC) and Spanish guidelines for laboratory animal care.