Article Text

Abstract
Chronic lung diseases represent a major public health problem with only limited therapeutic options. An important unmet need is to identify compounds and drugs that target key molecular pathways involved in the pathogenesis of chronic lung diseases. Over the last decade, there has been extensive interest in investigating Wingless/integrase-1 (WNT) signalling pathways; and WNT signal alterations have been linked to pulmonary disease pathogenesis and progression. Here, we comprehensively review the cumulative evidence for WNT pathway alterations in chronic lung pathologies, including idiopathic pulmonary fibrosis, pulmonary arterial hypertension, asthma and COPD. While many studies have focused on the canonical WNT/β-catenin signalling pathway, recent reports highlight that non-canonical WNT signalling may also significantly contribute to chronic lung pathologies; these studies will be particularly featured in this review. We further discuss recent advances uncovering the role of WNT signalling early in life, the potential of pharmaceutically modulating WNT signalling pathways and highlight (pre)clinical studies describing promising new therapies for chronic lung diseases.
- Airway Epithelium
- Asthma
- COPD ÀÜ Mechanisms
- Cytokine Biology
- Idiopathic pulmonary fibrosis
- COPD Pharmacology
- Asthma Mechanisms
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- Airway Epithelium
- Asthma
- COPD ÀÜ Mechanisms
- Cytokine Biology
- Idiopathic pulmonary fibrosis
- COPD Pharmacology
- Asthma Mechanisms
The basics: WNT signalling
The Wingless/integrase-1 (WNT) signalling pathways represent classical developmentally active pathways required for proper organ development. WNT ligands comprise a family of secreted glycoproteins that instruct cells in the respiratory system to adopt particular fates throughout lung development as well as tissue homoeostasis in adulthood. In humans, the WNT ligand family is composed of 19 distinct ligands, which are historically classified based on their amino acid sequence rather than their functional properties. Classically, WNT signalling has been separated into canonical and non-canonical signalling. WNT signalling that relies on the activation of the transcriptional coactivator β-catenin is designated as canonical WNT signalling (figure 1) and pathways activated by WNT ligands independently of β-catenin are classified as non-canonical WNT pathways (figure 2). We start off with an explanation of the well described canonical WNT signalling pathway, in which WNT ligands activate β-catenin-mediated gene transcription. In the absence of specific WNT ligands, cytosolic β-catenin is tightly regulated by the so-called ‘β-catenin destruction complex’, a multiprotein complex that targets β-catenin via phosphorylation and ubiquitination for proteasomal degradation (figure 1). The core of the ‘β-catenin destruction complex’ is composed of the proteins axin, adenomatous polyposis coli, casein kinase-1 and glycogen synthase kinase-3β (GSK-3β). The latter is the primary kinase involved in the phosphorylation and subsequent degradation of β-catenin. Binding of a specific WNT ligand (eg, WNT-3A) to one of the Frizzled receptors (FZD1 through FZD10) and subsequent activation of the low-density lipoprotein receptor-related proteins 5 and 6 (LRP5/6) co-receptors triggers an intracellular signalling cascade, which results in inactivation of the ‘β-catenin destruction complex’. Hence, cytosolic β-catenin can accumulate, translocate to the nucleus and, in association with T cell factor/lymphoid enhancer factor-1 (TCF/LEF) family of transcription factors, induce specific gene expression (figure 1).1

Schematic representation of canonical WNT/β-catenin signalling. Left side: cytosolic β-catenin is rapidly degraded by the β-catenin destruction complex in the absence of extracellular WNT ligands. The core of the β-catenin destruction complex is composed of: adenomatous polyposis coli (APC), axin, casein kinase-1 (CK-1) and glycogen synthase kinase-3 (GSK-3). GSK-3 is the primary kinase involved in the degradation of β-catenin. Right side: an extracellular WNT ligand binds and activates Frizzled (FZD) and the low density lipoprotein receptor-related proteins 5 and 6 (LRP5/6), which results in the activation of and intercellular signalling cascade that leads to the inhibition of the β-catenin destruction complex. Hence, β-catenin can accumulate and translocate to the nucleus to induce gene transcription. In the nucleus β-catenin can associate with various transcriptional coactivators, including T cell factor (TCF) and lymphoid enhancer factor (LEF).

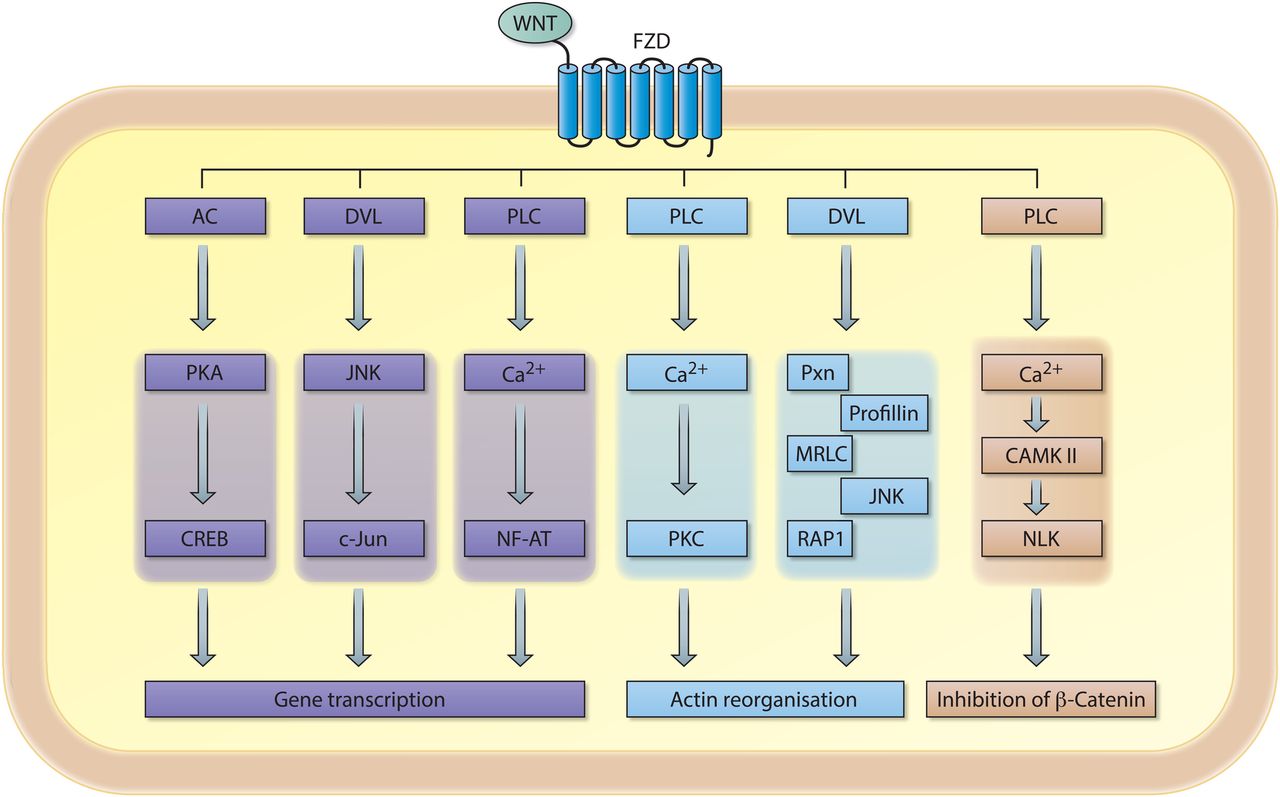
Schematic representation of signalling cascades involved in non-canonical WNT signalling. An extracellular WNT ligand binds to the Frizzled (FZD) receptor, which can subsequently activate a variety of downstream signalling cascades involved in gene transcription, intercellular actin organisation and/or inhibition of the transcriptional coactivator β-catenin. AC, adenylylcyclase; PKA, protein kinase A; CREB, cAMP responsive element binding protein; DVL, dishevelled; FZD, Frizzled receptor; JNK, c-Jun-N terminal kinase; PLC, phospolipase C; NF-AT, nuclear factor of activated T cells; PKC, protein kinase C; PXN, paxillin; MRLC, myosin regulatory light chain; RAP1, RAS-related protein 1; CAMKII, calcium/calmodulin-dependent kinase II; NLK, Nemo-like-kinase.
Activation of non-canonical WNT signalling also relies on the binding of specific WNT ligands (eg, WNT-4 or WNT-5A) to FZD receptors; however, it appears to be independent of LPR5/6 co-receptors. Non-canonical WNT signalling results in the activation of intracellular signalling molecules involved in planar cell polarity (PCP pathway), calcium/calmodulin-dependent protein kinase II (Ca2+/CAMKII) signalling and/or various less well defined downstream effector molecules (figure 2). Notably, some classically defined non-canonical WNT ligands are able to negatively influence canonical WNT/β-catenin signalling. Moreover, single WNT ligands can activate multiple signalling pathways suggesting that WNT ligands are not intrinsically canonical or non-canonical. Selectivity in receptor-ligand binding (eg, FZD-WNT interaction) likely dictates the outcome of downstream signalling.2 ,3 Indeed, a biochemical study demonstrated that WNT ligands can selectively bind to specific FZD receptors, and that respective WNT-FZD pairs exert functional selectivity in downstream signalling.4 These data emphasise the interconnectivity and complexity of canonical and non-canonical WNT signalling1 ,5–7 (figure 2). The dynamics of WNT and FZD expression in complex biological systems in vivo is currently unknown, thus a better understanding of receptor-ligand interactions in WNT signalling is required to decipher how exactly WNT ligands function. As such, the separation of WNT signalling in purely canonical and non-canonical signalling pathways appears to be outdated and certainly oversimplifies the complexity of this signalling pathway; however for uniformity reasons we maintain this nomenclature in this review. Whenever possible, we mention which WNT ligands, receptors and/or downstream signalling molecules are involved when we refer to canonical or non-canonical WNT signalling.
Over the last decade, there has been extensive interest in investigating WNT signalling pathways in chronic lung diseases. Several components of the WNT pathways serve as potent oncogenes and WNT signalling has been linked to lung cancer, which has been extensively reviewed previously and will not be included in this review.5 ,8–13 Here, we aimed to comprehensively review cumulative evidence for WNT pathway alterations in chronic lung pathologies, including idiopathic pulmonary fibrosis (IPF), pulmonary arterial hypertension (PAH), asthma and COPD. Early studies have largely focused on the canonical WNT/β-catenin signalling pathway and only recently several reports suggest that non-canonical WNT signalling might also contribute significantly to chronic lung pathologies. These studies will be highlighted in this review. We further discuss recent advances in our knowledge on the role of WNT signalling in early life, and feature novel developments and the potential application of WNT signalling modulation for drug development and (pre)clinical studies.
WNT signalling in early life
Despite intensive research efforts, the aetiology of major chronic lung diseases in children and adults remains elusive. Several lines of evidence indicate that prenatal and/or early postnatal lung injuries will have important implications for future lung function and increase risk for development of chronic lung diseases later in life.14 Several reports highlight the functional importance of canonical and non-canonical WNT signalling in lung morphogenesis and postnatal development and this has been reviewed previously.8 ,15–19 Ectopic expression of specific WNT ligands during lung development, either those involved in canonical or non-canonical signalling, can result in severe lung phenotypes, which partially resemble lung diseases observed during adulthood.5 ,20–23 Moreover, deletion of β-catenin in epithelial cells of embryonic lungs results in disrupted lung morphogenesis.24 In contrast, overexpression of a truncated, constitutively active form of β-catenin in Clara cell secretory protein (CCSP) positive cells (Club cells) does not influence lung morphogenesis before birth, but leads postnatally to goblet cell hyperplasia, pulmonary tumour development and airspace enlargement.25 Collectively, these studies highlight the importance of strict spatiotemporal control of WNT signalling for proper lung development and lung physiology early in life and thereafter into adulthood. More recently, studies have focused on the impact of environmental factors on prenatal and postnatal lung development. Epidemiological studies have shown that maternal smoking is a risk factor for the development of several chronic lung diseases.26 ,27 It was demonstrated in mice that maternal smoking negatively affects the mRNA expression of the WNT pathway components frizzled7 (Fzd7; receptor) and Ctnnb1 (gene symbol of β-catenin) as well as the WNT target gene Fn (fibronectin) in lung tissue of the offspring.28 Although not investigated in humans, impairment of these WNT pathway components may have implications for lung development, and fitness later in life, as these WNT components regulate neoangiogenesis and lung branching morphogenesis.28 Thus, the various WNT pathways are part of a signalling network essential for proper lung development and physiology, which may contribute to (age-associated) chronic lung diseases when impaired at early stages of life.29
WNT signalling in pulmonary fibrosis
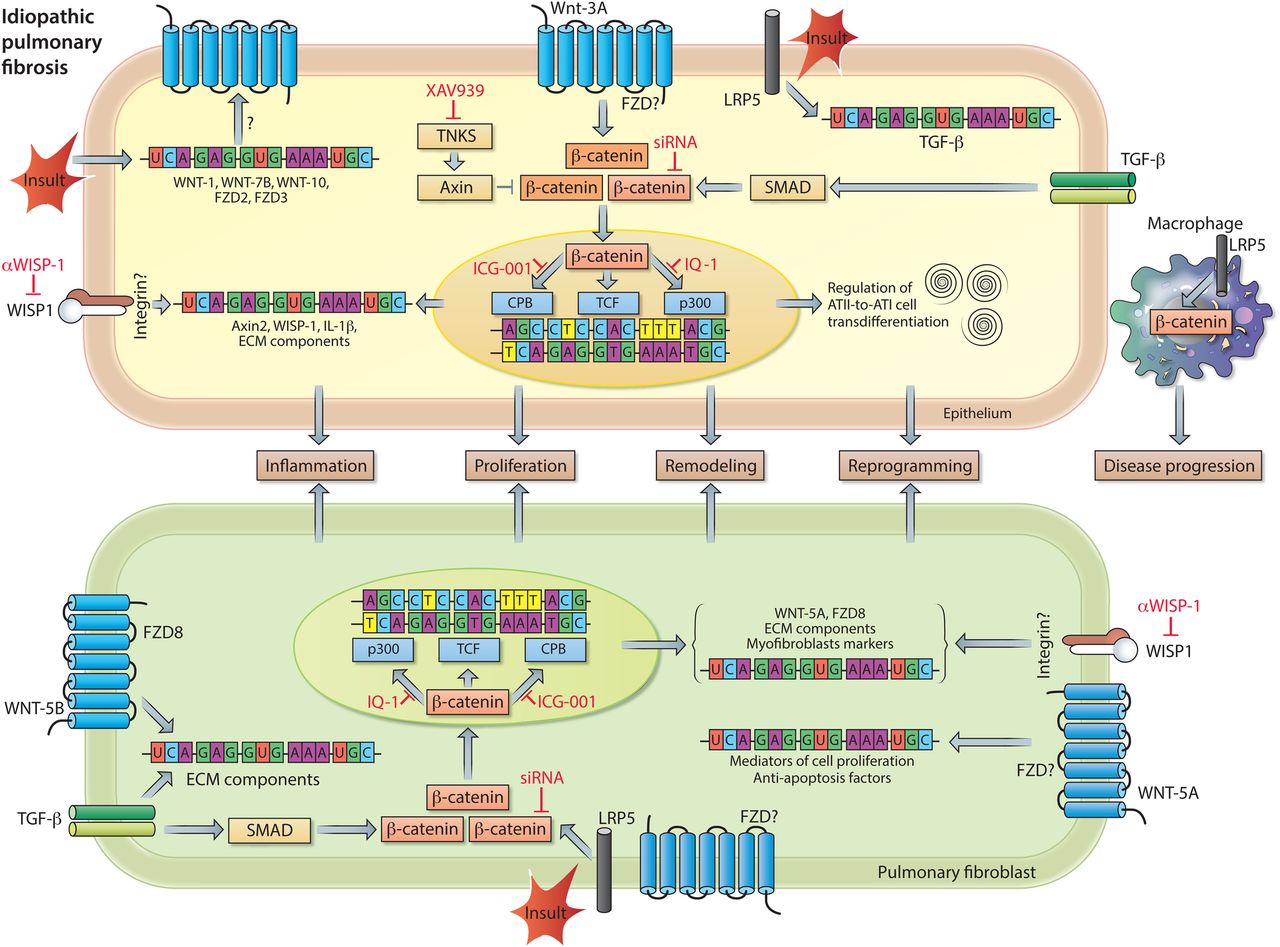
Impairment of the WNT signalling pathways and their potential role as therapeutic targets in chronic lung disease was first identified and established in IPF. IPF is a devastating, progressive disease characterised by lung epithelial injury and reprogramming, fibroblasts activation and excessive extracellular matrix (ECM) remodelling resulting in distorted lung architecture and a progressive loss of functional lung tissue.30 Although our understanding of IPF pathogenesis has significantly improved over the recent years, we currently have only limited pharmacological approaches available to treat the disease.25 ,31 ,32 The concept that developmental pathways (such as WNT, Sonic Hedgehog, Notch pathways) are altered in IPF, arose largely from unbiased gene expression profiling of models of experimental lung fibrosis as well as human IPF lung tissue specimens, which particularly indicated a ‘WNT signature’ within the IPF lung.33–36 It has been proposed that IPF lungs show remarkable resemblances to the developing lung in terms of pathology. This further implies that particular developmental signalling pathways may be reactivated in adult tissues following injury and contribute to IPF pathogenesis. Indeed, canonical WNT/β-catenin signalling is active in various cell types in human and experimental pulmonary fibrosis34 ,35 (figure 3). More specifically, increased gene expression of WNT-1, WNT-7B, WNT-10B, FZD2, FZD3, CTNNB1 (β-catenin) and LEF1 was observed in lung tissue of individuals with IPF compared with individuals without IPF (donor).34 Immunohistochemical analysis localised the classically defined canonical ligands WNT-1 and WNT-3A as well as the WNT effector protein β-catenin largely to bronchial and alveolar epithelium, although increased nuclear β-catenin expression has also been observed in (myo)fibroblasts in fibrotic foci.34 ,35 ,37 Increased expression of WNT target genes and, in particular, the increase in WNT1-inducible signalling protein-1 (WISP1) protein expression suggests that functional WNT/β-catenin signalling activity is enhanced in lung tissue of individuals with IPF.34 ,35 Antibody-mediated inhibition of WISP1 results in decreased lung pathology in bleomycin-induced lung fibrosis in mice in vivo and WISP1 has been reported to regulate alveolar epithelial cell function and reprogramming as well as (myo)fibroblast activation, thereby impairing lung function.35 Successive studies demonstrated that WISP1 is a WNT target gene, and at the same time a common downstream mediator of several profibrotic factors, including (1) miR-92A, a micro-RNA that is downregulated in human and experimental lung fibrosis, and (2) Transforming growth factor β (TGFβ) and Tumor necrosis factor α (TNFα) signalling in human lung fibroblasts, in which WISP1 regulates cell proliferation in an interleukin 6 (IL-6)-dependent fashion.38 ,39 Altogether, these findings corroborate WISP1 as a therapeutic target in IPF. With regard to upstream WNT/β-catenin signalling; extensive research has focused on preventing excessive activation of β-catenin signalling by targeting this developmental pathway at various molecular levels (figure 3). Several studies demonstrate that pharmacological and genetic inhibitors of β-catenin signalling attenuate fibrosis in various organs, including the lung.40–46 Noteworthy, activation of β-catenin in alveolar epithelial cells at the early stages of injury may reflect an attempt to repair and regenerate, however sustained activation of the pathway may drive inflammation and fibrotic changes in the lung.9 ,42 ,47–53
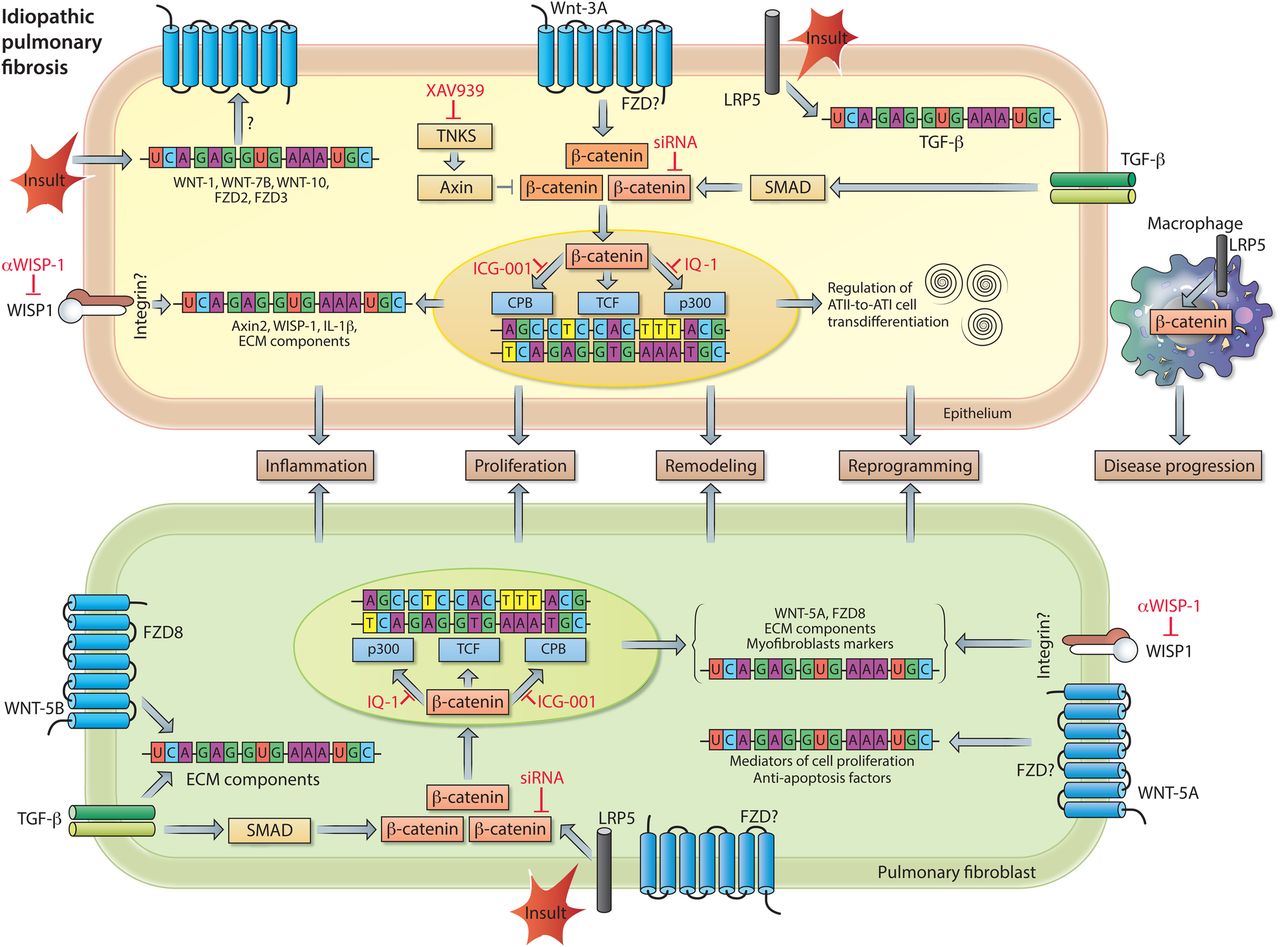
WNT signalling in idiopathic pulmonary fibrosis (IPF) pathogenesis. Increased pulmonary expression of WNT-1, WNT-7B, WNT-10B, Frizzled receptor (FZD)2 and FZD3 in individuals with IPF. Enhanced expression of transcriptionally active β-catenin in pulmonary epithelial cells (top), as a consequence of WNT-3A and/or TGF-β signalling. β-catenin signalling induces mRNA expression of inflammatory and remodelling markers (eg, IL-1β and WNT1-inducible signalling protein-1 (WISP1)) and regulates alveolar epithelial type II cells (ATII)-to-ATI-cell transdifferentiation, a process implicated in wound healing and tissue regeneration. The profibrotic action of WISP1 can be diminished by neutralising antibodies. In pulmonary fibroblasts (bottom), WNT-5B by activating FZD8, in conjunction with TGF-β signalling, causes upregulation of mRNA expression of other WNT signal components, extracellular matrix (ECM) components, and myofibroblast markers. Both the expression of ECM components and markers of myofibroblast differentiation are dependent on activation of transcriptionally active β-catenin. Accumulation of transcriptionally active β-catenin can be prevented by small molecule inhibitors of Tankyrases (eg, XAV939), whereas the interaction of β-catenin with specific transcription factors can be inhibited by ICG-001 (β-catenin/cAMP response element-binding protein binding protein (CBP)), PKF115–584 (β-catenin/TCF (T cell factor)) or IQ-1 (β-catenin/p300). WNT-5A, via a yet unknown FZD, induces proliferation and protects cells from oxidative-stress-induced apoptosis. Lipoprotein receptor-related protein 5 (LRP5) and TGF-β signalling are indispensable for activation of β-catenin signalling in response to a fibrotic insult. Moreover, LRP5 in macrophages contributes to disease progression. See main text for further details.
The alveolar epithelium, consisting of alveolar epithelial type I and II cells (ATI and ATII cells, respectively), represents a major site of tissue damage during lung injury. ATII cells are capable of self-renewal and exert progenitor function for ATI cells upon alveolar epithelial injury, a process dependent on autocrine WNT/β-catenin signalling.48 ,50 ,54 ,55 In physiological conditions, mature ATII cells exhibit a low degree of constitutive β-catenin signalling in vivo; however, the pathway is activated when the lungs are subjected to bleomycin-induced injury.48 ,50 Activation of the WNT/β-catenin signalling pathway is also observed during ATII-to-ATI cell transdifferentiation in vitro. The transdifferentiation process is characterised by increased expression of several WNT ligands, enhanced phosphorylation of WNT signalling intermediates (ie, LRP6 and DVL3), increased transcriptionally active β-catenin and an increase in β-catenin-driven target gene expression.48 ,50 Collectively, these data indicate that β-catenin activation during alveolar epithelial cell transdifferentiation is dependent on endogenous, autocrine canonical WNT signalling. This was confirmed by experiments in which Dickkopf-1 (DKK-1), an endogenous inhibitor of canonical WNT signalling, was overexpressed. These experiments demonstrated that DKK-1 attenuates β-catenin-driven target gene expression (ie, Axin2) in transdifferentiating ATII cells.48 Furthermore, inhibition of canonical WNT signalling by either the small molecule PKF115–584 (disruption β-catenin/TCF interaction), β-catenin siRNA or by ectopic expression of inhibitor of β-catenin and TCF (ICAT) attenuates the time-dependent increase in ATI cell marker expression in these primary cell cultures.48 ,50 Ectopic expression of ICAT also impairs wound closure after scratch-induced injury.48 Taken together, these data indicate that WNT/β-catenin signalling regulates alveolar epithelial cell transdifferentiation and repair processes, which might initially be beneficial upon lung injury. Nevertheless, persistent WNT/β-catenin activation in the alveolar epithelium may be detrimental in fibrotic lung diseases, like IPF. An unbiased gene expression screen to identify cell-specific mediators of canonical WNT signalling in primary murine ATII cells identified the proinflammatory cytokine IL-1β as one of the highest upregulated genes induced by WNT-3A stimulation.47 Subsequent experiments confirmed increased protein level of IL-1β in ATII cells in response to WNT-3A, as well as in an animal model of experimental lung fibrosis and, most importantly, in human IPF. These findings indicate that the alveolar epithelium is a relevant source of proinflammatory/profibrotic cytokines induced by active WNT/β-catenin signalling in pulmonary fibrosis.47 Among many tested, only four cytokines/growth factors (ie, IL-1β, TNFα, Granulocyte macrophage colony stimulating factor (GM-CSF) and TGF-β) have thus far been identified to be able to drive lung fibrosis.56 Transient overexpression of IL-1β initially results in inflammatory response with tissue injury followed by a progressive fibrotic response mediated in part by TGF-β.57 Thus, sustained canonical WNT signalling activity in alveolar epithelial cells may contribute via IL-1β to lung fibrosis. Furthermore, alveolar epithelial cell reprogramming and epithelial-to-mesenchymal transition (EMT) potentially contributes to chronic lung diseases, like IPF or COPD.58–62 TGF-β is the most commonly used growth factor for promoting epithelial cell reprogramming and its interaction with WNT signalling is important for cell fate determination during development and in the adult. Zhou et al,63 reported that TGF-β induces the expression of the mesenchymal marker α-smooth muscle-actin (α-sm-actin) with concomitant upregulation of transcriptionally active β-catenin in lung epithelial cells. Inhibition of β-catenin diminished TGF-β-induced α-sm-actin expression in these cells, whereas enhanced activation of β-catenin via GSK-3β inhibition (ie, LiCl) augmented this process.63 Subsequent experiments showed that α-sm-actin is transcriptionally regulated by a complex consisting of SMAD3/β-catenin/cAMP response element-binding protein binding protein (CBP).63 Furthermore, Ulsamer et al 45showed that the EMT process, characterised by increased expression of collagen, α-sm-actin and the transcription factor twist, is largely dependent on activation of a specific form of β-catenin (ie, pY654-β-catenin). Accumulation of pY654-β-catenin can be attenuated in vitro and in vivo by inhibition of tankyrase-1 (TNKS1), which partially prevents the mesenchymal changes both in primary alveolar epithelial cells and in an animal model of lung fibrosis.45 Tankyrases (TNKS1 and 2, respectively) are a class of enzymes that facilitate Axin proteolysis, which results in decreased assembly of the ‘β-catenin destruction complex’ and subsequently increases β-catenin signalling.1 ,64 Hence, inactivation of tankyrases attenuates canonical WNT/β-catenin signalling. The therapeutic potential of using tankyrase inhibitors in established experimental lung fibrosis was examined by Wang et al.46 Administration of the tankyrase inhibitor XAV939 starting 10 days after the administration of bleomycin to the mice results in attenuation of β-catenin signalling, improved survival of the mice and a significant reduction in lung pathology.46 Thus, inhibition of this family of enzymes is of potential interest to target β-catenin signalling and to reduce profibrotic responses in the lung. The advantageous effects of XAV939 may be attributed to reduced fibroblasts proliferation, impaired myofibroblast differentiation and, potentially, increased differentiation of bone marrow-derived mesenchymal stem cells into epithelial-like cells.46 However, more extensive in vitro and in vivo analyses still have to be performed to unambiguously support the latter finding. Nevertheless, this combined body of evidence suggests that the large repertoire of β-catenin regulated genes is decisive of cellular fate. In the nucleus, gene transcription induced by β-catenin is facilitated by the recruitment and activation of various factors, including transcription factors, proteins involved in histone methylation and acetylation (ie, MLL1/2, CBP and p300), histone deacetylases and histone modifiers (eg, BRG1).65 β-Catenin as a transcriptional-coactivator has a plethora of binding partners and its interaction with distinct transcription factors (eg, TCFs, SMADs, forkhead box (FOXO) or HIF1α) has important implications for its transcriptional activity and determines the functional output.66–68 One elegant and promising therapeutic option for IPF is to ‘re-direct’ β-catenin signalling by using specific inhibitors that target the interaction of β-catenin with distinct transcriptional cofactors. This approach has been applied in a study by Henderson et al 42 in which the authors demonstrated that selective inhibition of the β-catenin/CBP interaction by the ICG-001 led to reversal of established bleomycin-induced pulmonary fibrosis and improved epithelial cell integrity, whereas this was not achieved by the food and drugs administration (FDA) approved drug pirfenidone. Based on these results it was proposed that the interaction of β-catenin with CBP drives gene transcription critical for the maintenance of cells in an undifferentiated/proliferative state, whereas the interaction of β-catenin with p300 (a transcriptional coactivator homologous to CBP) initiates cellular differentiation. A subsequent study by Rieger et al 69 dissected the role of p300 and CBP in adult progenitor cell differentiation focusing on ATII-to-ATI cell transdifferentiation. Transdifferentiation of rat ATII cells to ATI-like cells was dependent on the interaction of β-catenin with p300, but independent of the interaction with CBP. Pharmacological or genetic disruption of the β-catenin/p300 interaction resulted in decreased expression of aquaporin5 (AQP5; ATI cell marker) and partial stabilisation of surfactant protein C (ATII cell marker) expression. Interestingly, the interaction between β-catenin and p300 in this context was facilitated by the classically defined non-canonical WNT ligand WNT-5A via a molecular mechanism involving protein kinase C-mediated phosphorylation of p300.69 This study and various other studies demonstrate that targeting β-catenin-dependent signalling may be beneficial for progenitor cell function and consequently the treatment of fibrosis. The question remains if and which secreted WNT ligands actually represent the driving force for the activation of β-catenin in human and experimental pulmonary fibrosis. A better understanding and determination of (cell)-specific ligand/receptor complexes is of high interest, as it will foster the identification of potential therapeutic targets. To elucidate if canonical WNT ligands contribute to disease pathogenesis, Lam et al 44 investigated the role of the indispensable WNT co-receptors LRP5 and LRP6 in murine and human pulmonary fibrosis. Notably, LRP5 transcript levels were associated with IPF progression, disease severity at presentation, and negatively correlated with clinical parameters like diffusion capacity of the lung for carbon monoxide (DLco) and composite physical index (CPI). This study furthermore demonstrated that LRP5-deficient fibroblasts exhibit reduced capacity to activate β-catenin and that mice lacking LRP5 (LRP5−/−mice) were protected against bleomycin-induced lung fibrosis.44 In a subsequent study by the same group, microarray analysis revealed that LRP5−/− mice that were exposed to bleomycin showed enrichment for pathways related to ECM processing and the innate immune response, suggesting that the immune cell-ECM remodelling axis is important in fibrosis. Macrophage-specific deletion of β-catenin did not have an effect on the development of bleomycin-induced lung fibrosis; however, the resolution of fibrosis in this model was accelerated.70 Similarly, in a murine model of asbestos-induced, non-resolving lung fibrosis was shown that loss of LRP5 does not influence development of fibrosis, but does result in delayed progression of fibrosis.70 These data indicate that macrophages with activated WNT/β-catenin signalling play an important role in sustaining pulmonary fibrosis.
In addition to LRP5/6, only a few WNT receptors have been investigated thus far in chronic lung diseases. The previously mentioned study also reported high FZD8 expression in individuals with IPF, which correlated to more rapid disease progression.44 Indeed, cellular signalling mediated via FZD8 is involved in fibrotic responses in the lung.71 FZD8 is the most profound upregulated FZD receptor in primary human lung fibroblasts in response to TGF-β stimulation. Moreover, genetic silencing of FZD8, in part, prevents the profibrotic action of TGF-β in pulmonary fibroblasts in vitro and in bleomycin-induced lung fibrosis in vivo. Importantly, profibrotic signalling via FZD8 appears to be mediated by WNT-5B and is independent of β-catenin signalling.71 These findings suggest that non-canonical WNT signalling also contributes to IPF pathogenesis. In line with this, increased WNT-5A expression has been demonstrated in IPF.72–74 Vuga et al,74 reported that this non-canonical WNT ligand mediates ECM deposition by pulmonary fibroblasts and protects these cells against oxidative stress-induced apoptosis, thereby potentially contributing to IPF. Collectively, cumulative evidence strongly suggests that the developmental WNT signalling pathways are able to drive fibrotic changes in the lung and are of great therapeutic interest for the treatment of IPF (figure 3).
WNT signalling in Pulmonary arterial hypertension
PAH is a well recognised complication of interstitial lung diseases like IPF, although PAH can also develop without underlying parenchymal lung disease(s). Both idiopathic PAH (IPAH), as well as common acquired forms of PAH, are characterised by a progressive increase in pulmonary vascular resistance, due to (1) loss of peripheral pulmonary arteries as a consequence of endothelial cell apoptosis and (2) occlusion of larger proximal arteries by increased smooth muscle cell proliferation and enhanced ECM deposition. The pathomechanisms in PAH are multifactorial and not fully understood, but include chronic hypoxia and hypoxic vasoconstriction, mechanical lung stress, smoking effects and inflammation.75 Aberrant growth factor signalling has also been implicated in the pathogenesis of PAH. Heterozygous loss of function mutations in bone morphogenetic protein receptor II (BMPRII) is observed in both sporadic and familial cases of IPAH.76 ,77 These findings imply that dysfunction of the developmentally active Bone morphogenetic protein (BMP) signalling is involved in PAH development. Interestingly, BMP-2 via BMPRII recruits canonical and non-canonical WNT pathways to promote pulmonary arterial endothelial proliferation, survival and migration.78 Activation of canonical WNT/β-catenin signalling, as well as non-canonical WNT/RHOA/RAC1 signalling, are necessary for vascular growth in vivo, suggesting that WNT signalling pathways are required for BMP-2-mediated angiogenesis and therefore might be beneficial in PAH.78 Nevertheless, an independent study investigating genetic signatures common across multiple cell lines that are associated with pathological processes in PAH identified WNT signalling as a target pathway, which was validated further in vitro. 79 The data from this study suggest that alterations in WNT signalling are a common molecular defect in both heritable PAH and IPAH, which is linked to decreased BMPRII signalling.79 Several components of the PCP pathway (non-canonical WNT signalling) are upregulated in pulmonary resistance vessels in IPAH.80 Accordingly, WNT ligand expression (eg, WNT-5A) positively correlated to pulmonary arterial pressure in pulmonary arterial smooth muscle cells from patients with PAH.81 Furthermore, Wu et al 82 investigated the expression and molecular target(s) of several microRNAs in the pathogenesis of PAH and discovered that Mir-199b-5p was overexpressed in PAH and negatively regulated the expression of GSK-3β resulting in enhanced β-catenin-driven gene transcription. These findings suggest enhanced WNT signalling in PAH. Thus, WNT signalling pathways have a crucial role in the regulation of pulmonary angiogenesis and vascular remodelling, and therapies that modulate WNT pathway activity could be of use in patients with PAH. The complexity of WNT pathway signalling, the extent of crosstalk with other (growth factor-dependent) signalling pathways and the plethora of signalling components, remain challenges to identify safe and effective therapies that specifically target WNT signalling in PAH.
WNT signalling in asthma
Asthma is a chronic airway disease with high prevalence in children as well as adults. The hallmark pathological features of asthma include eosinophilic airway inflammation and structural changes within the airways (airway remodelling), which are associated with an irreversible loss in lung function.83 Identifying genetic determinants for lung function is important in providing insight into the pathophysiology of asthma and other chronic lung diseases. One of the first studies suggesting a genetic link between WNT signalling and asthma was performed by Sharma et al,84 who demonstrated that WNT signalling genes are associated with impaired lung function in asthmatic children with WISP1 associating with FEV1 and FVC, whereas WNT inhibitory factor-1 (WIF-1) associated with FVC and FEV1/FEV (not with FEV1 alone). Furthermore, a single nucleotide polymorphism (SNP; rs2929973) in the WISP1 gene is significantly associated with FEV1 in asthmatic children, whereas an SNP in WIF-1 does influence lung function (ie, FEV1).85 Moreover, a recent publication, which used a genome-wide association study (GWAS)-based analysis to prioritise asthma susceptibility genes, revealed two biological pathways to be enriched in asthma pathogenesis: (1) cytokine-cytokine receptor interactions and (2) WNT signalling pathway(s). The same study identified a novel susceptibility locus near WNT signalling genes, in particular SNPs located near FZD3 and FZD6.86
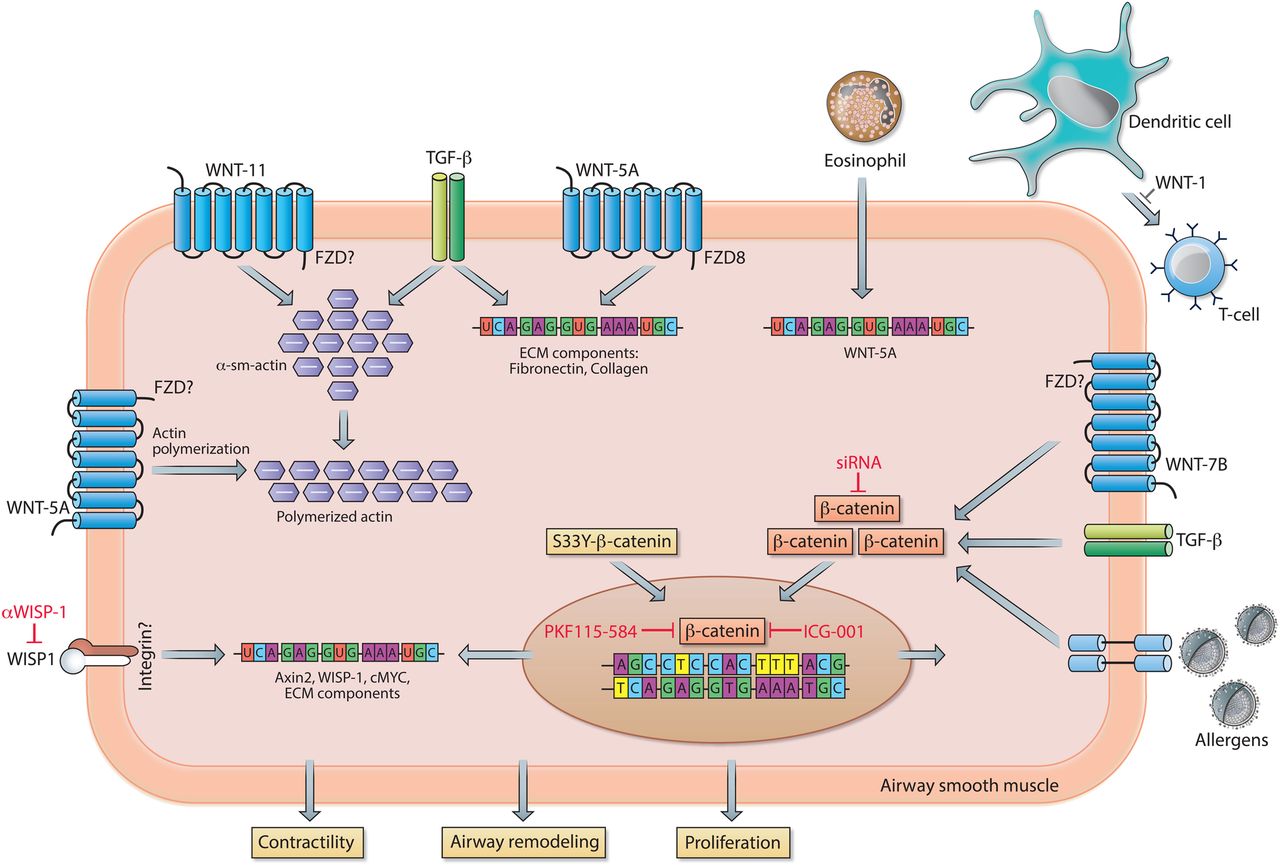
Airway inflammation in allergic asthma is mainly characterised by a T helper 2 cell (Th2) signature and eosinophilia.87 In human asthmatic airways, gene expression of multiple WNT ligands is positively (WNT-3A, WNT-5A, WNT-6 and WNT-10A) or negatively (WNT-5B) associated with a Th2 signature. In addition, FZD5 (WNT receptor) is upregulated in patients with Th2-high asthma.87 Recently, a study investigated whether eosinophil-induced airway remodelling in asthma is associated with alterations in WNT expression. Eosinophils derived from patients with asthma enhanced the expression of WNT-5A, TGF-β and gene expression of several ECM components in human airway smooth muscle cells. In addition, these eosinophils induced airway smooth muscle cell proliferation, which is potentially mediated by WNT-5A. This study links WNT signalling to inflammatory processes, and airway remodelling; however, this was not directly addressed experimentally.88 Furthermore, these studies suggest that WNT signalling potentially modulates allergic responses in the lung. By using the mouse model of ovalbumin (OVA)-induced allergic asthma, Reuter et al 89 examined the impact of WNT-1 on development of allergic lung disease. Overexpression of this specific WNT ligand in CCSP-positive cells (Club cells) had beneficial effects on allergic airway disease, demonstrated by a reduction in airway hyper-responsiveness (AHR), decreased eosinophilia in the bronchoalveolar lavage and a reduced number of mucus cells in comparison to animals that did not overexpress the ligand. The beneficial effects of WNT-1 expression were due to impairment of dendritic cell-dependent activation of T cells.89 In addition to inflammation, the impact of WNT signalling on airway remodelling has been investigated using animal models of allergic asthma, in which increased expression of specific WNT ligands (ie, Wnt-5A and Wnt-7B), β-catenin and the canonical WNT target gene Axin2 was observed.90 ,91 Genetic silencing of β-catenin, by siRNA, attenuated inflammation and airway remodelling in asthmatic mice in vivo. 90 Airway remodelling may be, in part, a consequence of a process in which pulmonary epithelial cells acquire a mesenchymal phenotype (EMT). In accordance, the aeroallergen house dust mite, together with TGF-β, induces mesenchymal transition of bronchial epithelial cells. This process was associated with loss of E-cadherin, a binding partner of β-catenin at the plasma membrane. This potentially leads to dissociation of β-catenin from the membrane and the subsequent translocation of β-catenin to the nucleus, where it assists the transcription of mesenchymal genes.92 Furthermore, in a rat model of allergic asthma, airway remodelling in response to OVA exposure was partially prevented by antibody-mediated inhibition of the WNT target gene WISP1.93 In a more recent study, no differences were observed in β-catenin expression in the airway smooth muscle layer in either an animal model of allergic asthma or in smooth muscle biopsies from asthmatic individuals compared with non-asthmatic donors.94 β-Catenin-driven gene expression (Axin2, Wisp1 and c-Myc), however, was highly increased in the used mouse model of OVA-induced asthma. Moreover, pharmacological inhibition of β-catenin/CBP interaction by the small molecule ICG-001 prevented smooth muscle remodelling and ECM expression in in vitro and in vivo models of asthma.94 In agreement, β-catenin signalling is required for TGF-β-induced expression of ECM proteins by human airway smooth muscle cells and pulmonary fibroblasts. Furthermore, enhanced synthesis of the ECM protein fibronectin by human airway smooth muscle cells was observed after ectopic expression of constitutively active β-catenin (ie, S33Y-β-catenin; resistant to GSK-3-mediated proteasomal degradation).95 ,96 On the other hand, while overexpression of a non-degradable form of β-catenin (S37A-β-catenin) in fibroblasts is insufficient to induce TGF-β, profibrotic marker expression and/or myofibroblast differentiation, it does increase fibroblast migration and proliferation.43 Of note, β-catenin might further play an important role in airway smooth muscle proliferation and contraction independently of WNT signalling. The latter is presumably due to its role in cell-cell contacts as a component of cell adherens junctions.97 ,98 This may represent an additional mechanism by which β-catenin contributes to asthma pathology. By and large, these findings indicate that β-catenin is a potential therapeutic target in asthma, which is discussed in more detail in a comprehensive review by Kumawat et al.17
Remarkably, among all the WNT ligands detected in human airway smooth muscle cells, WNT-5A is the most abundant WNT ligand expressed.99 Moreover, WNT-5A protein expression is approximately twofold higher in airway smooth muscle of asthmatics compared with healthy donors. These data suggest a potential role for non-canonical WNT signalling as well in asthma.100 Stimulation of airway smooth muscle cells with solely WNT-5A was not sufficient to induce ECM deposition; however, siRNA-mediated silencing of WNT-5A largely blunted TGF-β-induced ECM deposition (collagen 1α1 and fibronectin) by airway smooth muscle cells.99 Furthermore, subsequent in vitro studies demonstrated that WNT-5A as well as WNT-11 may contribute to AHR by inducing actin polymerisation and the upregulation of α-sm-actin in human airway smooth muscle, respectively.101 ,102 Whether this pathway is also involved in asthma pathogenesis in vivo is not yet elucidated. In summary, these data indicate that specific WNT ligands, by acting on various cell types, potentially contribute to pathogenic processes in asthma, including airway inflammation and remodelling, and highlight (canonical WNT-driven) β-catenin as a potential therapeutic target in asthma.
WNT signalling in COPD
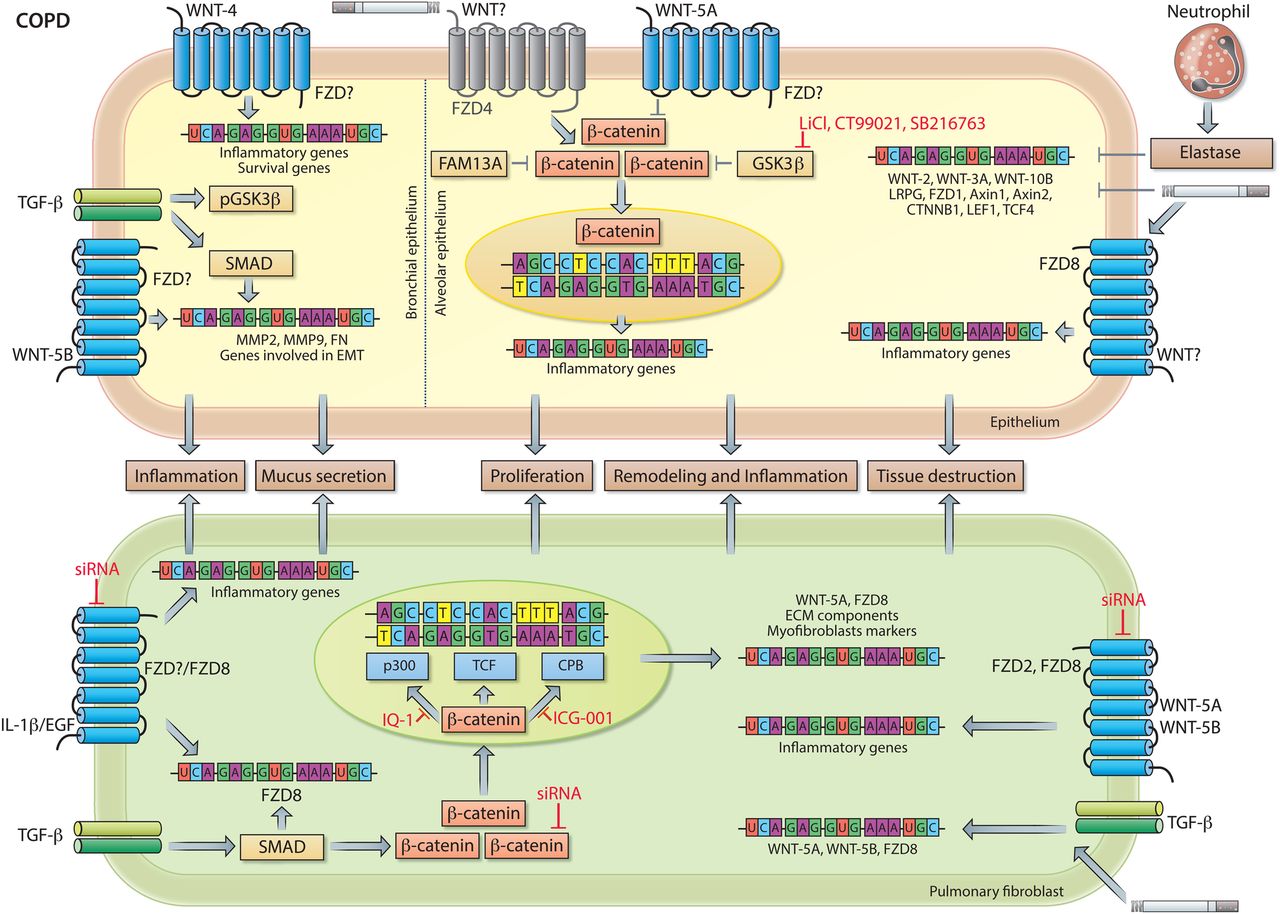
COPD is characterised by chronic airflow limitation caused by (1) small airway disease, which is composed of (small) airway remodelling and chronic bronchitis, and (2) parenchymal destruction, which leads to emphysema. The relative contribution of each of these pathological features varies in individual patients who suffer from COPD. Given the importance for WNT/β-catenin signalling for lung development and growth, this pathway represents a prime target for initiation of lung repair and regeneration. A tremendous challenge in emphysema is the restoration of functional lung tissue and one current concept is that the progressive destruction of tissue is partially due to the inability of the lung to activate self-repair mechanisms in COPD. Similarly to IPF, no therapy is currently available for COPD that stabilises or reverses disease progression.103 ,104 Cigarette smoke, the main risk factor for developing COPD, reduces canonical WNT signalling in vitro and in vivo in human bronchial epithelial cells.105 ,106 Other studies report that bronchial epithelial cells of smokers and individuals with COPD might exhibit enhanced nuclear β-catenin expression and that components of cigarette smoke (eg, nicotine) can induce EMT in a WNT-3A/β-catenin-dependent manner.62 ,107 Nevertheless, transcript levels of several components of canonical WNT signalling (eg, CTNNB1, GSK-3β and TCF4) are significantly lower in peripheral lung tissue of individuals with COPD compared with smokers without COPD, indicating that processes involved in COPD pathogenesis affect components of canonical WNT signalling independently of smoking.105 These data confirm previous findings in which expression profiles obtained from lung tissue specimens from smokers without obstruction and individuals with COPD were compared.108 In this study, several biological pathways that may be relevant to COPD pathogenesis independently of smoking were identified, including TGF-β signalling, proteins involved in focal adhesion and WNT signalling pathways.108 Notably, in experimental and human COPD/emphysema canonical WNT signalling is silenced in the alveolar epithelium as observed by decreased nuclear expression of β-catenin, a surrogate marker for canonical WNT signalling activity (figure 5).109 Several recent studies investigated potential underlying mechanism(s) for reduced β-catenin signalling in COPD.110–112 family with sequence similarity 13 member A (FAM13A) was identified in a GWAS as a gene that predisposes to increased susceptibility to COPD.113 ,114 A subsequent study investigated the functional role of FAM13A in COPD pathogenesis and demonstrated that FAM13A induces emphysema by targeting canonical WNT signalling.111 Increased expression of FAM13A was observed in several cell types in the lungs of patients with COPD and FAM13A-deficient mice (FAM13A−/−) were protected against cigarette smoke-induced as well as elastase-induced emphysema development. The resistance to elastase-induced emphysema in FAM13A−/− mice was abrogated by coadministration of a β-catenin inhibitor (PKF118–310). Biochemical experiments revealed that FAM13A associates with protein phosphatase 2A to facilitate GSK-3β-mediated degradation of β-catenin. Collectively, this study demonstrated that FAM13A is increased in lungs of patients with COPD and might contribute to COPD susceptibility by promoting β-catenin degradation.111 An extensive analysis of FZD receptor expression in experimental and human COPD, identified FZD4 as an important regulator of canonical WNT/β-catenin signalling and potential candidate for therapeutic intervention in this chronic lung disease.112 In physiological conditions, the FZD4 receptor is expressed on alveolar epithelial cells, however the expression of this receptor is diminished in individuals with moderate and very severe COPD (Global initiative for chronic obstructive lung diseases (GOLD) stages II and III/IV). Moreover, FZD4 expression correlated positively to lung function (%FEV1, postbronchodilator) and negatively with smoking pack-years in the analysed cohort of patients. In vivo and in vitro studies demonstrated that cigarette smoke was able to directly attenuate FZD4 expression, which was accompanied by a reduction in active β-catenin expression.112 This suggests that FZD4 is a positive regulator of β-catenin signalling in alveolar epithelial cells. Pharmacological inhibition of FZD4 via the small molecule FzM1 decreased (WNT-3A-driven) β-catenin signalling in alveolar epithelial cells, whereas overexpression of FZD4 had the opposite effect. Functional studies demonstrated that the receptor is important for wound healing and repair by the alveolar epithelium, as FZD4 facilitates cell proliferation, migration and ATII-to-ATI cell transdifferentiation.112 Taken together, these findings strongly suggest that FZD4 signalling and β-catenin activation might be a promising route to induce lung repair in COPD. Indeed, preventive as well as therapeutic reactivation of β-catenin signalling via GSK-3β inhibition resulted in alveolar epithelial cell activation, attenuated emphysema pathology, and improved lung function in experimental emphysema in vivo. 109 Notably, the potential therapeutic application of WNT/β-catenin activation in COPD has recently been tested in more detail and translated into human tissue specimen using three-dimensional lung tissue cultures (3D-LTCs).115 Patient-derived 3D-LTCs are generated ex vivo and represent a valuable tool for preclinical target validation and drug testing. Using this set-up, we demonstrated the potential of activating canonical WNT/β-catenin signalling in 3D-LTCs derived from lung tissue of individuals with COPD.115 Activation of β-catenin by GSK-3β inhibition, by either LiCl or CT99021, led to increased alveolar epithelial cell marker expression, decreased matrix metalloproteinase-12 expression, as well as altered macrophage activity and elastin remodelling in 3D-LTCs.115 Altogether, these data underline the potential suitability of WNT/β-catenin activators, such as GSK-3β inhibitors, for the treatment of COPD and urge further preclinical studies for this devastating disease (figure 5).
Similarly, to IPF and asthma, WNT signalling independent of β-catenin (non-canonical WNT signalling) may contribute to COPD pathogenesis. Two independent studies nearly simultaneously showed aberrant WNT-4 expression in bronchial cells of patients with COPD.116 ,117 This specific WNT ligand induces cell proliferation and proinflammatory cytokine secretion by bronchial epithelial cells. In addition, two structurally closely related WNT ligands, WNT-5A and WNT-5B, have been recently investigated in COPD.96 ,110 ,118 ,119 WNT-5B is predominantly expressed in airway epithelial cells with significantly higher WNT-5B staining in bronchial epithelial cells of patients with COPD compared with smokers.118 In vitro experiments with recombinant WNT-5B revealed that this specific WNT ligand induces expression of genes related to airway remodelling (ie, FN, MMP-2 and MMP-9) independently of β-catenin signalling, but required activation of TGF-β/SMAD3 signalling.118 In addition, WNT-5A has also been linked to inflammatory processes by pulmonary fibroblasts and COPD pathogenesis. Individual components of cigarette smoke, including nicotine, can induce WNT-5A in cells of the respiratory system and WNT-5A is a direct target of miR-487b, a microRNA that is repressed by cigarette smoke.110 ,120 ,121 Moreover, cigarette smoke can directly activate TGF-β, a growth factor implicated in WNT-5A induction in several distinct cells of the lungs.72 ,96 ,99 ,100 ,110 These findings indicate WNT-5A as a prime target of COPD-related exposures. Most recently, we reported increased WNT-5A transcript and protein in lung tissue of individuals with COPD compared with non-COPD controls.110 We furthermore observed altered post-translational modifications (ie, glycosylation, palmitoylation and oligomerisation) of WNT-5A in lung tissue specimens of individuals with COPD compared with tissue of donors.110 The increased expression of this ligand was recapitulated in a murine model of cigarette smoke-induced COPD as well as in elastase-induced emphysema.110 Notably, various studies have indicated that specific non-canonical WNT ligands are able to inhibit canonical WNT/β-catenin signalling.3 ,122 Accordingly, mature WNT-5A attenuated canonical WNT/β-catenin-driven alveolar epithelial cell wound healing and ATII-to-ATI cell transdifferentiation in vitro. Furthermore, lung-specific overexpression of WNT-5A aggravated elastase-induced emphysema in vivo. Most importantly, both prophylactic as well as therapeutic inhibition of WNT-5A, using neutralising antibodies or a synthetic peptide, resulted in attenuated tissue destruction, improved lung function and restoration of β-catenin-driven transcriptional targets (eg, Axin2 and Nkd1) as well as alveolar epithelial cell marker expression in two animal models of COPD.110 These findings suggest a canonical to non-canonical WNT signal switch in COPD, which contributes to disease development and progression. Moreover, our study highlights that non-canonical WNT signalling and, in particular targeting WNT-5A, is a promising therapeutic option for the treatment of emphysema/COPD.110 However, which FZD receptors are mediating the effects of these WNT ligands (ie, WNT-4, WNT-5A and/or WNT-5B) in COPD is largely unknown. Nevertheless, a recent study by Spanjer et al 123 linked the WNT receptor FZD8 to chronic bronchitis and mucus hypersecretion, but did not investigate which WNT ligand(s) might be involved in these processes, and this needs further examination.
In summary, it appears that canonical and non-canonical WNT signalling pathways play opposing or balancing roles in COPD, wherein (re)activation of WNT/β-catenin signalling is a potential remedy for emphysema whereas non-canonical WNT signalling contributes to airway remodelling and cigarette smoke-induced inflammation, thus perpetuating disease development and progression (figure 5).
Fighting the cold WNT-er
An important unfulfilled need is to identify new agents that interact with key molecular pathways involved in the pathogenesis of chronic lung diseases. Since the last decade, our knowledge about deviations in WNT signalling in the aforementioned chronic lung disease continues to grow (figures 3 ⇓–5 and table 1). The ability to target the WNT signalling pathway offers immense promise in chronic lung diseases; however, substantial risks and concerns remain present with regard to the targeting of such crucial signalling pathways. The challenge for the future will be identifying which branch of WNT signalling to target at what time point and how to specifically target major signalling hubs within these pathways. Inhibition of biogenesis and secretion of WNT ligands as well as blocking the interaction of WNT ligands with the cell-surface receptors can be achieved to a certain extent by pharmacologically active small molecules.124 While these approaches do not necessarily discriminate between targeting either the canonical or non-canonical WNT signalling pathway and are therefore relatively non-specific, this may also be a potential benefit as both these WNT signalling pathways are involved in pathological processes in IPF and asthma. A major benefit of the canonical WNT signalling pathway over the non-canonical pathways is that β-catenin is a specific downstream effector molecule of canonical signalling, which can be targeted pharmacologically. Activation of β-catenin signalling is of therapeutic potential in emphysema/COPD and can be achieved by various molecular mechanisms. Most commonly, β-catenin signalling is activated via inhibition of GSK-3β, the primary kinase involved in β-catenin degradation.1 Prudence is advised with artificially activating β-catenin signalling in the lung due to the risk of potential adverse effects, such as EMT, tissue remodelling or tumorigenesis. However, in a guinea pig model of lipopolysaccharide (LPS)-induced COPD airway wall remodelling was substantially reduced by pharmacological inhibition of GSK-3β.125 Moreover, the GSK-3β inhibitor lithium has been used to treat patients for many years now without apparent increases in the cancer incidence.126 Taken together, these studies suggest that inhibition of GSK-3β has therapeutic potential in COPD. In contrast to emphysema/COPD, increased β-catenin signalling is a hallmark of IPF and inhibition of this transcriptional coactivator is of great therapeutic interest. Most pharmacological approaches to attenuate canonical WNT/β-catenin signalling focus on blocking the interaction of β-catenin with the transcription factors.127 A primary consideration, however, is that β-catenin-dependent signalling can also be activated by other growth factors than WNTs and that transcriptional activity of β-catenin is influenced by its interaction with various binding partners.
Overview of molecular targets and applied tools/compounds to modify WNT signalling, which have been investigated in chronic lung diseases

WNT signalling in asthma. In airway smooth muscle cells, WNT-5A activates Frizzled receptor (FZD)8, which together with TGF-β stimulation results in increased mRNA expression of extracellular matrix (ECM) components. In addition, WNT-5A, via a not further specified FZD, enhances actin polymerisation and contractile capacity of the smooth muscle cell. Eosinophil-driven airway inflammation stimulates the smooth muscle cells to increase WNT-5A expression. Increased expression of WNT-1 prevents dendritic cell-mediated activation of T cells, thereby attenuating airway hyper-responsiveness (AHR) and airway remodelling. WNT-11, via an unspecified FZD, conjointly with TGF-β stimulation causes upregulation of the contractile protein α-sm-actin. Activation of transcriptionally active β-catenin in response to WNT-7B, TGF-β and/or (aero)allergens results in augmented mRNA expression of ECM components and genes involved in cell proliferation. Similarly, ectopic expression of a non-degradable form of β-catenin (S33Y-β-catenin) enhances expression of ECM components. Transcriptional activity of β-catenin can be inhibited by PKF115–584 or ICG-001, whereas the canonical WNT target gene WNT1-inducible signalling protein-1 (WISP1) can be inhibited with neutralising antibodies. See main text for further details.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
WNT signalling in COPD. Neutrophil elastase and cigarette smoke attenuate pulmonary expression of WNT-2, WNT-3A, WNT-10B, LRP6, FZD1, AXIN1, AXIN2, CTNNB1 (β-catenin), LEF1 and TCF4 in human and/or animal models of COPD. In bronchial epithelial cells (left side of dashed line), WNT-4, independently of β-catenin, induces expression of extracellular matrix (ECM) components and of genes involved in cell proliferation. WNT-5B together with TGF-β/SMAD signalling activates gene transcription of MMP2, MMP9 and FN. Additionally, TGF-β inactivates glycogen synthase kinase-3 (GSK-3)β via phosphorylation resulting in activation of β-catenin, which facilitates the epithelial-to-mesenchymal transition (EMT) process of bronchial epithelial cells. In alveolar epithelial cells (right side of dashed line), β-catenin is a critical regulator of ATII-to-ATI-cell transdifferentiation. WNT-5A, which is increased in individuals with COPD and secreted by pulmonary fibroblasts, acts a negative regulator of β-catenin signalling, thereby impairing endogenous tissue repair by alveolar epithelial cells. FAM13, a COPD susceptibility gene, together with GSK-3β contributes to the development of emphysema by enhanced targeting of β-catenin for proteasomal degradation in alveolar epithelial cells. Moreover, cigarette smoke inhibits β-catenin signalling and epithelial cell repair by reducing FZD4 expression (indicated in grey). Pharmacological reactivation of β-catenin signalling via GSK-3β inhibition (by eg, LiCl, CT99021 or SB216763) in experimental emphysema in vivo as well as patient-derived COPD tissue ex vivo results in epithelial cell activation and attenuated emphysema pathology. In pulmonary fibroblasts, expression of proinflammatory genes induced by IL-1β or Epidermal growth factor (EGF) is mediated in part by FZD8. Additionally, IL-1β and EGF induce the expression of FZD8 via a yet unidentified signalling cascade, whereas TGF-β-induced expression of FZD8 is dependent on SMAD signalling. Additionally, TGF-β induces the expression of WNT-5A and WNT-5B, which in turn induce expression of proinflammatory genes in a FZD2-dependent and/or FZD8-dependent manner. Activated WNT signalling together with TGF-β induces mRNA expression of WNT signalling components, ECM components and myofibroblast markers. β-Catenin is required for the expression of ECM components and myofibroblast differentiation. See main text for further details.
Finally, the majority of chronic lung diseases affect diverse cellular compartments, and cell-specific targeting of WNT signalling to ensure a beneficial clinical outcome represents a challenge for the future. Our knowledge about (cell-)specific ligand/receptor complexes has just recently begun to increase and this route holds promise for treatment applications, as drugs targeting distinct WNT ligands and/or FZD receptors are currently under investigation.1 ,128–132 Taken together, several valuable approaches to target the WNT pathways exist or are under development and thus offer encouraging routes to potentially treat chronic lung diseases in the future.
Acknowledgments
The authors thank all members of the Königshoff laboratory, in particular Florian Ciolek, Stephan Klee, Mareike Lehmann, Carlo Mümmler, Wioletta Skronska-Wasek and Darcy Wagner, for stimulating discussions about the content and the title of this review at ATS 2016.
References
Footnotes
Twitter Follow Melanie Königshoff @m_konigshoff
Contributors HAB and MK conceptually designed and wrote the manuscript.
Funding HAB is supported by a postdoctoral fellowship from the European Respiratory Society (ERS Fellowship LTRF 79-2012) and a fellowship from Helmholtz Zentrum Germany (PFP PF-135). MK is supported by a European Research Council Starting Grant (ERC-StG-LS7, Grant No. 261302) and a W2/3 Professorship Award by the Helmholtz Association.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.